Dosage Form (Pharmaco-Technical) Factors
| Home | | Biopharmaceutics and Pharmacokinetics |Chapter: Biopharmaceutics and Pharmacokinetics : Absorption of Drugs
1. Disintegration Time 2. Manufacturing/Processing Variables 3. Pharmaceutical Ingredients/Excipients (Formulation factors) 4. Nature and Type of Dosage Form 5. Product Age and Storage Conditions
DOSAGE FORM (PHARMACO-TECHNICAL) FACTORS
1. Disintegration Time
2. Manufacturing/Processing Variables
3. Pharmaceutical Ingredients/Excipients (Formulation factors)
4. Nature and Type of Dosage Form
5. Product Age and Storage Conditions
Disintegration Time
Disintegration time (DT) is of particular importance in case of solid dosage forms like tablets and capsules. In vitro disintegration test is by no means a guarantee of drug’s bioavailability because if the disintegrated drug particles do not dissolve, absorption is not possible. However, if a solid dosage form does not conform to the DT, it portends bioavailability problems because the subsequent process of dissolution will be much slower and absorption may be insufficient. Coated tablets, especially sugar coated ones have long DT. Rapid disintegration is thus important in the therapeutic success of a solid dosage form. DT of a tablet is directly related to the amount of binder present and the compression force (hardness) of a tablet. A harder tablet with large amount of binder has a long DT. Disintegration can be aided by incorporating disintegrants in suitable amounts during formulation.
After disintegration of a solid dosage form into granules, the granules must deaggregate into fine particles, as dissolution from such tiny particles is faster than that from granules.
Manufacturing/Processing Variables
Drug dissolution is the single most important factor in the absorption of drugs, especially from the most widely used conventional solid dosage forms, tablets and capsules. The dosage form related factors that influence dissolution and hence absorption of a drug from such formulations are:
1. Excipients (formulation ingredients apart from the active principles), and
2. Manufacturing processes.
The influence of excipients such as binders, lubricants, disintegrants, etc. on drug dissolution will be discussed in the subsequent section of this chapter.
Several manufacturing processes influence drug dissolution from solid dosage forms. Processes of such importance in the manufacture of tablets are:
1. Method of granulation, and
2. Compression force.
The processing factor of importance in the manufacture of capsules that can influence its dissolution is the intensity of packing of capsule contents.
Method of Granulation: The wet granulation process is the most conventional technique in the manufacture of tablets and was once thought to yield tablets that dissolve faster than those made by other granulation methods. The limitations of this method include—
(i) Formation of crystal bridge by the presence of liquid,
(ii) The liquid may act as a medium for affecting chemical reactions such as hydrolysis, and
(iii) The drying step may harm the thermolabile drugs.
Involvement of large number of steps each of which can influence drug dissolution—method and duration of blending, method, time and temperature of drying, etc.
The method of direct compression has been utilized to yield tablets that dissolve at a faster rate. One of the more recent methods that have resulted in superior product is agglomerative phase of communition (APOC). The process involves grinding of drugs in a ball mill for time long enough to affect spontaneous agglomeration. The tablets so produced were stronger and showed rapid rate of dissolution in comparison to tablets made by other methods. The reason attributed to it was an increase in the internal surface area of the granules prepared by APOC method.
Compression Force: The compression force employed in tabletting process influence density, porosity, hardness, disintegration time and dissolution of tablets. The curve obtained by plotting compression force versus rate of dissolution can take one of the 4 possible shapes shown in Fig. 2.25.
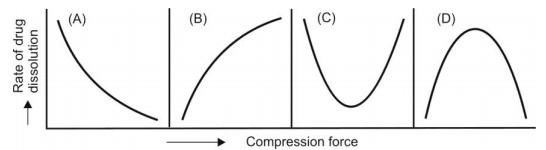
Fig. 2.25. Influence of compression force on dissolution rate of tablets
On the one hand, higher compression force increases the density and hardness of tablet, decreases porosity and hence penetrability of the solvent into the tablet, retards wettability by forming a firmer and more effective sealing layer by the lubricant, and in many cases, promotes tighter bonding between the particles, all of which result in slowing of the dissolution rate of tablets (curve A of Fig. 2.25). On the other hand, higher compression forces cause deformation, crushing or fracture of drug particles into smaller ones or convert a spherical granule into a disc shaped particle with a large increase in the effective surface area. This results in an increase in the dissolution rate of the tablet (curve B of Fig. 2.25). A combination of both the curves A and B is also possible as shown in curves C and D. In short, the influence of compression force on the dissolution rate is difficult to predict and a thorough study on each formulation should be made to ensure better dissolution and bioavailability.
Intensity of Packing of Capsule Contents: Like the compression force for tablets, packing density in case of capsule dosage form can either inhibit or promote dissolution. Diffusion of GI fluids into the tightly filled capsules creates a high pressure within the capsule resulting in rapid bursting and dissolution of contents. Opposite is also possible. It has been shown that capsules with finer particles and intense packing have poor drug release and dissolution rate due to a decrease in pore size of the compact and poor penetrability by the GI fluids.
Pharmaceutical Ingredients/Excipients (Formulation factors)
A drug is rarely administered in its original form. Almost always, a convenient dosage form to be administered by a suitable route is prepared. Such a formulation contains a number of excipients (non-drug components of a formulation). Excipients are added to ensure acceptability, physicochemical stability during the shelf-life, uniformity of composition and dosage, and optimum bioavailability and functionality of the drug product. Despite their inertness and utility in the dosage form, excipients can influence absorption of drugs. The more the number of excipients in a dosage form, the more complex it is and greater the potential for absorption and bioavailability problems. Commonly used excipients in various dosage forms are vehicles, diluents (fillers), binders and granulating agents, disintegrants, lubricants, coatings, suspending agents, emulsifiers, surfactants, buffers, complexing agents, colorants, sweeteners, crystal growth inhibitors, etc.
Vehicle: Vehicle or solvent system is the major component of liquid orals and parenterals. The 3 categories of vehicles in use are—aqueous vehicles (water, syrup, etc.), nonaqueous water miscible vehicles (propylene glycol, glycerol, sorbitol) and nonaqueous water immiscible vehicles (vegetable oils). Bioavailability of a drug from vehicles depends to a large extent on its miscibility with biological fluids. Aqueous and water miscible vehicles are miscible with the body fluids and drugs from them are rapidly absorbed. Quite often, a drug is more soluble in water miscible vehicles like propylene glycol (serving as a co-solvent) and show better bioavailability. Sometimes dilution of such vehicles with the body fluids results in precipitation of drug as fine particles which, however, dissolve rapidly. Solubilisers such as polysorbate 80 are sometimes used to promote solubility of a drug in aqueous vehicles. In case of water immiscible vehicles, the rate of drug absorption depends upon it’s partitioning from the oil phase to the aqueous body fluids, which could be a rate-limiting step. Viscosity of the vehicles is another factor in the absorption of drugs. Diffusion into the bulk of GI fluids and thus absorption of a drug from a viscous vehicle may be slower.
Diluents (Fillers): Diluents are commonly added to tablet (and capsule) formulations if the required dose is inadequate to produce the necessary bulk.
A diluent may be organic or inorganic. Among organic diluents, carbohydrates are very widely used—for example, starch, lactose, microcrystalline cellulose, etc. These hydrophilic powders are very useful in promoting the dissolution of poorly water-soluble, hydrophobic drugs like spironolactone and triamterene by forming a coat onto the hydrophobic surface of drug particles and rendering them hydrophilic. Among the inorganic diluents, dicalcium phosphate (DCP) is most common. One classic example of drug-diluent interaction resulting in poor bioavailability is that of tetracycline and DCP. The cause is formation of divalent calcium-tetracycline complex which is poorly soluble and thus, unabsorbable.
Binders and Granulating Agents: These materials are used to hold powders together to form granules or promote cohesive compacts for directly compressible materials and to ensure that the tablet remains intact after compression. Popular binders include polymeric materials (natural, semisynthetic and synthetic) like starch, cellulose derivatives, acacia, PVP, etc. Others include gelatin and sugar solution. In general, like fillers, the hydrophilic (aqueous) binders show better dissolution profile with poorly wettable drugs like phenacetin by imparting hydrophilic properties to the granule surface. However, the proportion of strong binders in the tablet formulation is very critical. Large amounts of such binders increase hardness and decrease disintegration/dissolution rates of tablets. PEG 6000 was found to be a deleterious binder for phenobarbital as it forms a poorly soluble complex with the drug. Non-aqueous binders like ethyl cellulose also retard drug dissolution.
Disintegrants: These agents overcome the cohesive strength of tablet and break them up on contact with water which is an important prerequisite to tablet dissolution. Almost all the disintegrants are hydrophilic in nature. A decrease in the amount of disintegrant can significantly lower bioavailability.
Adsorbing disintegrants like bentonite and veegum should be avoided with low dose drugs like digoxin, alkaloids and steroids since a large amount of dose is permanently adsorbed and only a fraction is available for absorption. Microcrystalline cellulose is a very good disintegrant (and a binder too) but at high compression forces, it may retard drug dissolution.
Lubricants/Antifrictional Agents: These agents are added to tablet formulations to aid flow of granules, to reduce interparticle friction and sticking or adhesion of particles to dies and punches. The commonly used lubricants are hydrophobic in nature (several metallic stearates and waxes) and known to inhibit wettability, penetration of water into tablet and their disintegration and dissolution. This is because the disintegrant gets coated with the lubricant if blended simultaneously which however can be prevented by adding the lubricant in the final stage. The best alternative is use of soluble lubricants like SLS and carbowaxes which promote drug dissolution.
Coatings: In general, the deleterious effect of various coatings on drug dissolution from a tablet dosage form is in the following order:
Enteric coat > Sugar coat > Non-enteric film coat.
The dissolution profile of certain coating materials change on aging; e.g. shellac coated tablets, on prolonged storage, dissolve more slowly in the intestine. This can, however, be prevented by incorporating little PVP in the coating formulation.
Suspending Agents/Viscosity Imparters: Popular suspending agents are hydrophilic polymers like vegetable gums (acacia, tragacanth, etc.), semisynthetic gums (CMC, MC) and synthetic gums which primarily stabilize the solid drug particles by reducing their rate of settling through an increase in the viscosity of the medium. These agents and some sugars are also used as viscosity imparters to affect palatability and pourability of solution dosage forms. Such agents can influence drug absorption in several ways. The macromolecular gums often form unabsorbable complexes with drugs—for example, sodium CMC forms a poorly soluble complex with amphetamine. An increase in viscosity by these agents acts as a mechanical barrier to the diffusion of drug from the dosage form into the bulk of GI fluids and from GI fluids to the mucosal lining by forming a viscid layer on the GI mucosa. They also retard the GI transit of drugs.
Surfactants: Surfactants are widely used in formulations as wetting agents, solubilisers, emulsifiers, etc. Their influence on drug absorption is very complex. They may enhance or retard drug absorption either by interacting with the drug or the membrane or both.
Mechanisms involved in the increased absorption of drug by use of surfactants include:
1. Promotion of wetting (through increase in effective surface area) and dissolution of drugs e.g. polysorbate 80 with phenacetin.
2. Better membrane contact of the drug for absorption.
3. Enhanced membrane permeability of the drug.
The beneficial effects of surfactants have been observed at pre-critical micelle concentration levels. However, physiologic surfactants like the bile salts (anionic) and lysolecithin (nonionic) promote absorption of hydrophobic drugs like steroids, oil soluble vitamins and griseofulvin by their micellar solubilising property.
Decreased absorption of drug in the presence of surfactants has been suggested to be due to:
1. Formation of unabsorbable drug-micelle complex at surfactant concentrations above critical micelle concentration
2. Laxative action induced by a large surfactant concentration
Buffers: Buffers are sometimes useful in creating the right atmosphere for drug dissolution as was observed for buffered aspirin tablets. However, certain buffer systems containing potassium cations inhibit the drug absorption as seen with vitamin B2 and sulphanilamide. The reason attributed to it was the uptake of fluids by the intestinal epithelial cells due to which the effective drug concentration in the tissue is reduced and the absorption rate is decreased. Such an inhibitory effect of the various buffer cations on the drug transfer rate is in the following order:
K+ > NH4+ > Li+ > Na+ > TRIS+.
Hence, the buffer system for a salt of a drug should contain the same cation as the drug salt and introduce no additional cations.
Complexing Agents: Complex formation has been used to alter the physicochemical and biopharmaceutical properties of a drug. A complexed drug may have altered stability, solubility, molecular size, partition coefficient and diffusion coefficient. Basically, such complexes are pharmacologically inert and must dissociate either at the absorption site or following absorption into the systemic circulation.
Several examples where complexation has been used to enhance drug bioavailability are:
1. Enhanced dissolution through formation of a soluble complex e.g. ergotamine tartarate-caffeine complex and hydroquinone-digoxin complex.
2. Enhanced lipophilicity for better membrane permeability e.g. caffeine-PABA complex.
3. Enhanced membrane permeability e.g. enhanced GI absorption of heparin (normally not absorbed from the GIT) in presence of EDTA which chelates calcium and magnesium ions of the membrane.
Complexation can be deleterious to drug absorption due to formation of poorly soluble or poorly absorbable complex e.g. complexation of tetracycline with divalent and trivalent cations like calcium (milk, antacids), iron (haematinics), magnesium (antacids) and aluminium (antacids).
Reasons for poor bioavailability of some complexes are –
4. Failure to dissociate at the absorption site, and
5. Large molecular size of the complex that cannot diffuse through the cell membrane—for example, drug-protein complex.
Colorants: Even a very low concentration of water-soluble dye can have an inhibitory effect on dissolution rate of several crystalline drugs. The dye molecules get adsorbed onto the crystal faces and inhibit drug dissolution— for example, brilliant blue retards dissolution of sulphathiazole. Dyes have also been found to inhibit micellar solubilisation effect of bile acids which may impair the absorption of hydrophobic drugs like steroids. Cationic dyes are more reactive than the anionic ones due to their greater power for adsorption on primary particles.
Precipitation/Crystal Growth Inhibitors: When a significant increase in free drug concentration above saturation or equilibrium solubility occurs, it results in supersaturation which in turn lead to drug precipitation or crystallization. Precipitation or crystal growth inhibitors such as PVP, HPMC, PEG, PVA (polyvinylalcohol) and similar such hydrophilic polymers prevent or prolong supersaturation thus preventing precipitation or crystallization by –
1. Increasing the viscosity of vehicle.
2. Prevent conversion of a high-energy metastable polymorph into stable, less soluble polymorph.
3. Adsorbing on the faces of crystal and reduce crystal growth.
Nature and Type of Dosage Form
Apart from the proper selection of drug, clinical success often depends to a great extent on the proper selection of dosage form of that drug. For a given drug, a 2 to 5 fold or perhaps more difference could be observed in the oral bioavailability of a drug depending upon the nature and type of dosage form. Such a difference is due to the relative rate at which a particular dosage form releases the drug to the biological fluids and the membrane. The relative rate at which a drug from a dosage form is presented to the body depends upon the complexity of dosage form. The more complex a dosage form, greater the number of rate-limiting steps and greater the potential for bioavailability problems.
The rate at which a particular dosage form releases the drug following administration is given in Fig. 2.26.
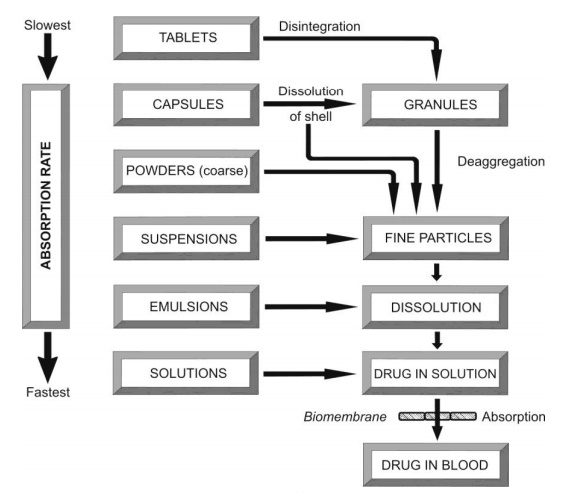
Fig. 2.26. Course of events that occur following oral administration of various dosage forms
As a general rule, the bioavailability of a drug from various dosage forms decreases in the following order:
Solutions > Emulsions > Suspensions > Capsules > Tablets > Coated Tablets > Enteric Coated Tablets > Sustained Release Products.
Thus, absorption of a drug from solution is fastest with least potential for bioavailability problems whereas absorption from a sustained release product is slowest with greatest bioavailability risk.
Several factors, especially the excipients which influence bioavailability of a drug from its dosage form, have been discussed earlier.
Solutions: A drug in a solution (syrups, elixirs, etc.) is most rapidly absorbed since the major rate-limiting step, drug dissolution, is absent. Factors that influence bioavailability of a drug from solution dosage form include—the nature of solvent (aqueous, water miscible, etc.), viscosity, surfactants, solubilisers, stabilizers, etc. Quite often, dilution of a drug in solution with GI fluids results in precipitation of drug as fine particles which generally dissolve rapidly. Factors that limit the formulation of a drug in solution form include stability, solubility, taste, cost of the product, etc.
Emulsions: Emulsion dosage forms have been found to be superior to suspensions in administering poorly aqueous soluble lipophilic drugs. It was observed with indoxole (an NSAID) that when it is dissolved in a vegetable oil and emulsified in water, absorption increases 3 fold over its aqueous suspension. Emulsion dosage form presents a large surface area of oil to the GIT for absorption of a drug. Scientists have claimed that a drug administered in oily vehicle (emulsified and solubilised in the GIT by bile salts to form mixed micelles) can direct the distribution of drug directly into the lymphatic system thereby avoiding the hepatic portal vein and first-pass metabolism.
Suspensions: The major rate-limiting step in the absorption of a drug from suspension dosage form is drug dissolution which is generally rapid due to the large surface area of the particles. Important factors in the bioavailability of a drug from suspensions include particle size, polymorphism, wetting agents, viscosity of the medium, suspending agents, etc.
Powders: Though powders are superior to tablets and capsules, they are not in use nowadays due to handling and palatability problems. Major factors to be considered in the absorption of a drug from powders are particle size, polymorphism, wettability, etc.
Capsules: Powders and granules are popularly administered in hard gelatin capsules whereas viscous fluids and oils in soft elastic shells. Factors of importance in case of hard gelatin capsules include drug particle size, density, polymorphism, intensity of packing and influence of diluents and excipients. Hydrophilic diluents like lactose improve wettability, deaggregation and dispersion of poorly aqueous soluble drugs whereas inhibitory effect is observed with hydrophobic lubricants like magnesium stearate. A hydrophobic drug with a fine particle size in capsule results in a decrease in porosity of powder bed and thus, decreased penetrability by the solvent with the result that clumping of particle occurs. This can be overcome by incorporating a large amount of hydrophilic diluent (upto 50%), a small amount of wetting agent cum lubricant such as SLS (upto 1%) and/or by wet granulation to convert an impermeable powder bed to the one having good permeability. Other factors of importance include possible interaction between the drug and the diluent (e.g. tetracycline-DCP) and between drug and gelatin shell. The influence of capsule processing factors on drug dissolution and bioavailability have already been discussed.
Soft elastic capsules as such dissolve faster than hard gelatin capsules and tablets and show better drug availability from oily solutions, emulsions or suspensions of medicaments (especially hydrophobic drugs). One of the best examples of this is the faster dissolution of indoxole (equivalent to that of an emulsion dosage form) when formulated as soft gelatin capsule in comparison to hard gelatin capsule and aqueous suspension. Such poorly soluble drugs can be dissolved in PEG or other suitable solvent with the aid of surfactants and encapsulated without difficulty. Soft gelatin capsules are thus of particular use where the drug dose is low, drug is lipophilic or when oily or lipid based medicaments are to be administered. A problem with soft gelatin capsules is the high water content of the shell (above 20%). This moisture migrates into the shell content and crystallization of drug occurs during the drying stage resulting in altered drug dissolution characteristics.
Tablets: Compressed tablets are the most widely used convenience and cost effective dosage forms. A schematic representation of disintegration, deaggregation, dissolution and absorption of a drug from a tablet dosage form is shown in Fig. 2.27.
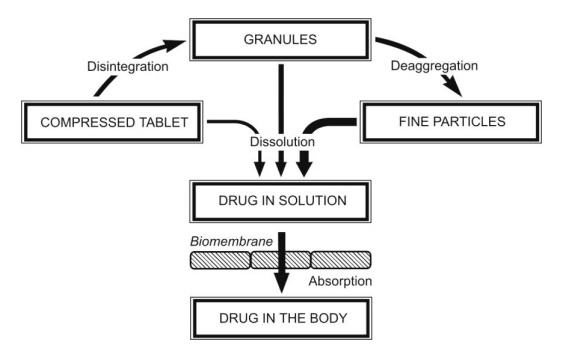
Fig. 2.27. Sequence of events in the absorption of a drug from tablet dosage form
The bioavailability problems with tablets arise from the reduction in the effective surface area due to granulation and subsequent compression into a dosage form. Since dissolution is most rapid from primary drug particles due to their large surface area, disintegration of a tablet into granules and subsequent deaggregation of granules into fine particles is very important. A number of formulation and processing factors influencing these steps and also the physicochemical properties of drug substance that influence bioavailability have already been discussed in the earlier sections of this chapter.
Coated Tablets: In addition to factors that influence drug release from compressed tablets, the coating acts as yet another barrier which must first dissolve or disrupt to give way to disintegration and dissolution of tablet. Of the two types of coatings, the film coat, which is thin, dissolves rapidly and does not significantly affect drug absorption. The sugar coat which though soluble, is generally tough and takes longer to dissolve. The sealing coat which is generally of shellac, is most deleterious. Press coated tablets may be superior to sugar coated tablets in such cases.
Enteric-Coated Tablets: Enteric coated tablets have great potential in creating bioavailability problems because the coat dissolves only in the alkaline pH of the intestine and it may take as long as 2 to 4 hours for such a tablet to empty from the stomach into the intestine depending upon the meals and the GI motility. Hence, the pharmacological response may eventually be delayed by as much as 6 to 8 hours. The problem of gastric emptying can, however, be overcome by enteric coating the granules or pellets and presenting them in a capsule or compressing into a tablet. The thickness of enteric coat is yet another determinant factor in drug dissolution, increasing thickness being more problematic. Aging of the dosage form also affects drug release, especially with shellac. In one of the studies, shellac coated tablets of salicylic acid stored for 2 years showed a 60% decrease in the peak plasma level.
Sustained-Release Products: Drug release from such products is most unpredictable, the problems ranging from dose dumping to unsatisfactory or no drug release at all. However, with the development of newer innovations and technologies, it is becoming increasingly reliable and the results reproducible with little inter-subject variations.
Product Age and Storage Conditions
A number of changes, especially in the physicochemical properties of a drug in dosage form, can result due to aging and alterations in storage conditions which can adversely affect bioavailability. With solution dosage form, precipitation of drug due to altered solubility, especially due to conversion of metastable into poorly soluble, stable polymorph can occur during the shelf-life of the product. Changes in particle size distribution have been observed with a number of suspension dosage forms resulting in decreased rate of drug dissolution and absorption. In case of solid dosage forms, especially tablets, disintegration and dissolution rates are greatly affected due to aging and storage conditions. An increase in these parameters of tablets has been attributed to excipients that harden on storage (e.g. PVP, acacia, etc.) while the decrease is mainly due to softening/crumbling of the binder during storage (e.g. CMC).
Changes that occur during the shelf-life of a dosage form are affected mainly by large variations in temperature and humidity. In one of the studies conducted on prednisone tablets containing lactose as the filler, high temperature and high humidity resulted in harder tablets that disintegrated and dissolved slowly.
Related Topics
