Non-Renal Routes of Drug Excretion
| Home | | Biopharmaceutics and Pharmacokinetics |Chapter: Biopharmaceutics and Pharmacokinetics : Excretion of Drugs
Drugs and their metabolites may also be excreted by routes other than the renal route, called as the extrarenal or nonrenal routes of drug excretion.
NON-RENAL ROUTES OF DRUG EXCRETION
Drugs and their metabolites may also be excreted by routes other than
the renal route, called as the extrarenal or nonrenal routes of drug excretion. The
various such excretion processes are:
1. Biliary excretion
2. Pulmonary excretion
3. Salivary excretion
4. Mammary excretion
5. Skin/dermal excretion
6. Gastrointestinal excretion
7. Genital excretion
1. Biliary Excretion of Drugs-Enterohepatic Cycling
The hepatic cells lining the bile canaliculi
produce bile. The production and secretion of bile are active processes. The
bile secreted from liver, after storage in the gall bladder, is secreted in the
duodenum. In humans, the bile flow rate is a steady 0.5 to 1 ml/min. Bile is
important in the digestion and absorption of fats. Almost 90% of the secreted
bile acids are reabsorbed from the intestine and transported back to the liver
for re-secretion. The rest is excreted in faeces.
Being an active process, bile secretion is
capacity-limited and subject to saturation. The process is exactly analogous to
active renal secretion. Different transport mechanisms exist for the secretion
of organic anions, cations and neutral polar compounds. A drug, whose biliary
concentration is less than that in plasma, has a small biliary clearance and vice versa. In some instances, the bile
to plasma concentration ratio of drug can approach 1000 in which cases, the
biliary clearance can be as high as 500 ml/min or more.
Compounds that are excreted in bile have been classified
into 3 categories on the basis of their bile/plasma concentration ratios:
Group A compounds whose ratio is
approximately 1, e.g. sodium, potassium and
chloride ions and glucose.
Group B compounds whose ratio is >1,
usually from 10 to 1000, e.g. bile salts, bilirubin glucuronide, creatinine, sulphobromophthalein conjugates, etc.
Group C compounds with ratio < 1, e.g.
sucrose, inulin, phosphates, phospholipids and mucoproteins.
Drugs can fall in any of the above three
categories.
Several factors
influence secretion of drugs in bile –
1. Physicochemical Properties of the Drug
The most important factor governing the excretion
of drugs in bile is their molecular weight. Its influence on biliary
excretion is summarized in the Table 6.3.
Polarity is the other physicochemical
property of drug influencing biliary excretion. Greater the polarity, better the excretion. Thus, metabolites are
more excreted in bile than the parent drugs because of their increased
polarity. The molecular weight threshold for biliary excretion of drugs is also
dependent upon its polarity. A threshold of 300 Daltons and greater than 300
Daltons is necessary for organic cations (e.g. quaternaries) and organic anions
respectively. Nonionic compounds should also be highly polar for biliary
excretion, e.g. cardiac glycosides.
TABLE 6.3
Influence of Molecular Weight on Excretion Behaviour of Drugs

2. Nature of Biotransformation Process
A metabolic reaction that greatly increases the
polarity as well as the molecular weight of drug favours biliary excretion of
the metabolite. Thus, phase II reactions, mainly glucuronidation and
conjugation with glutathione, result in metabolites with increased tendency for
biliary excretion (increase the molecular weight by 176 and 300 Daltons
respectively). Examples of drugs excreted in the bile as glucuronides are
morphine, chloramphenicol and indomethacin. Stilbestrol glucuronide is almost
entirely excreted in bile. Glutathione conjugates are exclusively excreted via
bile and are not observable in the urine because of their large molecular size.
Conjugation with amino acids and acetylation and methylation reactions do not
result in metabolites with greatly increased molecular weight and therefore
have little influence on biliary excretion of xenobiotics. For a drug to be
excreted unchanged in the bile, it must have a highly polar functional group
such as - COOH (cromoglycic acid), -SO3H (amaranth), -NH4+
(oxyphenonium), etc. Clomiphene citrate, an ovulation inducer, is almost completely
removed from the body via biliary excretion.
3. Other Factors
Miscellaneous factors influencing biliary excretion
of drugs include sex and species differences, protein-drug binding, disease
states, drug interactions, etc.
Substances having high molecular weight show good
excretion in bile in case of rats, dogs, and hen and poor excretion in rabbits,
guinea pigs and monkeys. The route is more important for the excretion of drugs
in laboratory animals than in man. Protein bound drugs can also be excreted in
the bile since the secretion is an active process. In cholestasis, the bile
flow rate is reduced thereby decreasing biliary excretion of drugs. Agents such
as phenobarbital stimulate biliary excretion of drugs, firstly, by enhancing
the rate of glucuronidation, and secondly, by promoting bile flow. The route of
drug administration also influences biliary drug excretion. Orally administered
drugs which during absorption process go to the liver, are excreted more in
bile in comparison to parenterally administered drugs. Food also has a direct
influence on biliary excretion of drugs. Protein and fat rich food increase
bile flow.
The efficacy of drug excretion by the biliary
system and hepatic function can be tested by an agent that is exclusively and
completely eliminated unchanged in the bile, e.g. sulphobromophthalein. This
marker is excreted within half an hour in the intestine when the hepatic
function is normal. A delay in its excretion is indicative of hepatic and
biliary malfunction. The marker is also useful in determining hepatic blood
flow rate.
The ability of liver to excrete the drug in the bile is expressed by biliary clearance (equation 6.24).
Biliary clearance = Biliary clearance rate / Plasma
drug concentration (6.24a)
= ( Bile flow x Biliary drug clearance ) / Plasma drug concentration (6.24b)
Just as the major portion of bile salts excreted in
intestine is reabsorbed, several drugs which are excreted unchanged in bile are
also absorbed back into the circulation. Some drugs which are excreted as
glucuronides or as glutathione conjugates are hydrolysed by the intestinal or
bacterial enzymes to the parent drugs which are then reabsorbed. The reabsorbed
drugs are again carried to the liver for resecretion via bile into the
intestine. This phenomenon of drug cycling between the intestine and the liver
is called as enterohepatic cycling or
enterohepatic circulation of drugs (Fig.
6.5.).
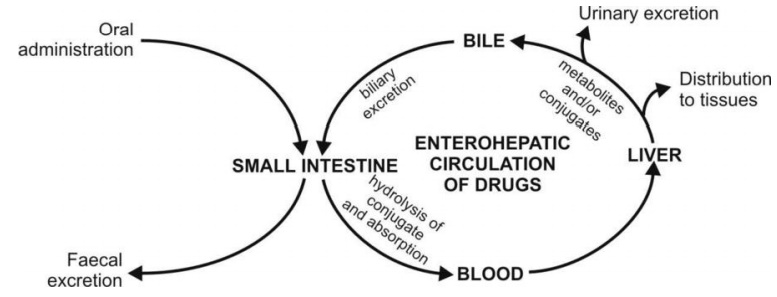
Fig. 6.5. Enterohepatic cycling of drugs
Such a recycling process continues until the drug
is biotransformed in the liver or is excreted in the urine or both. The drugs
which are secreted via bile in the intestine but not reabsorbed, are finally
excreted in the faeces.
Enterohepatic circulation is important in the conservation
of important endogenous substances such as vitamin B12, vitamin D3,
folic acid, several steroid hormones and bile salts. The process results in
prolongation of half-lives of several drugs (e.g. carbenoxolone) which are
extensively excreted in bile. The half-life of agents such as DDT, which are
resistant to biotransformation and are highly lipophilic, may increase to
several days due to such a recycling phenomenon. The prolonged therapeutic
activity of oral contraceptives (upto 12 hours) is also due to such a
recirculation. Other examples of drugs undergoing enterohepatic circulation are
cardiac glycosides, rifampicin, chlorpromazine and indomethacin. Drug
interactions affecting enterohepatic cycling occur when agents such as
antibiotics kill the intestinal microflora and thus retard hydrolysis of drug
conjugates and their subsequent reabsorption, or the unabsorbable ion exchange
resins such as cholestyramine which bind strongly to the acidic and neutral
drugs (e.g. digitoxin) and thus prevent their reabsorption. The principle of
adsorption onto the resins in the GIT can however be used to treat pesticide
poisoning by promoting their faecal excretion.
Biliary excretion of drugs can be assessed by
giving the drugs parenterally and detecting their presence in faeces. This also
rules out the doubt about the incomplete absorption of such drugs when given
orally and observed in faeces.
2. Pulmonary Excretion
Gaseous and volatile substances such as the general
anaesthetics (e.g. halothane) are absorbed through the lungs by simple
diffusion. Similarly, their excretion by diffusion into the expired air is
possible. Factors influencing pulmonary excretion of a drug include pulmonary
blood flow, rate of respiration, solubility of the volatile substance, etc. Gaseous
anaesthetics such as nitrous oxide which are not very soluble in blood are
excreted rapidly. Generally intact gaseous drugs are excreted but metabolites
are not. Compounds like alcohol which have high solubility in blood and tissues
are excreted slowly by the lungs. The principle involved in the pulmonary
excretion of benzene and halobenzenes is analogous to that of steam
distillation.
3. Salivary Excretion
Excretion of drugs in saliva is also a passive
diffusion process and therefore predictable on the basis of pH-partition
hypothesis. The pH of saliva varies from 5.8 to 8.4. The mean salivary pH in
man is 6.4. Unionised, lipid soluble drugs at this pH are excreted passively in
the saliva. Equations analogous to 6.3, 6.5, 6.6 and 6.7 can be written for drugs
with known pKa at the salivary pH and percent ionisation and
saliva/plasma drug concentration ratio (S/P) can be computed.
for weak acids,
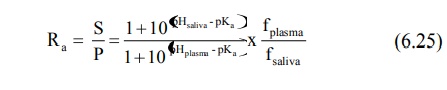
for weak bases,
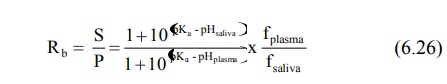
where, fplasma and fsaliva
are free drug fractions in plasma and in saliva respectively.
The S/P ratios have been found to be less than 1
for weak acids and greater than 1 for weak bases i.e. basic drugs are excreted
more in saliva as compared to acidic drugs. The salivary concentration of some
drugs reaches as high as 0.1%. Since the S/P ratio is fairly constant for
several drugs, their blood concentration can be determined by detecting the
amount of drug excreted in saliva, e.g. caffeine, theophylline, phenytoin,
carbamazepine, etc. Some drugs are actively secreted in saliva, e.g. lithium,
the concentration of which is sometimes 2 to 3 times that in plasma. Penicillin
and phenytoin are also actively secreted in saliva.
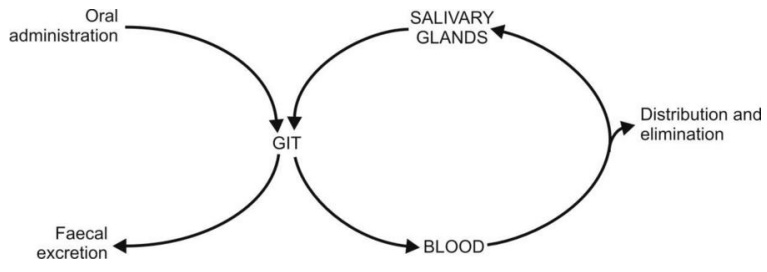
Fig. 6.6. Salivary cycling of drugs
The bitter after taste in the mouth of a patient on
medication is an indication of drug excretion in saliva. In few instances, the
process is responsible for side effects such as black hairy tongue in patients receiving
antibiotics, gingival hyperplasia due to phenytoin, etc.
Some basic drugs inhibit saliva secretion and are
responsible for dryness of mouth.
Drugs excreted in saliva can undergo cycling in a
fashion similar to enterohepatic cycling, e.g. sulphonamides, antibiotics,
clonidine, etc. (Fig. 6.6.).
4. Mammary Excretion
Excretion of a drug in milk is important since it
can gain entry into the breast-feeding infant.
Milk consists of lactic secretions originating from
the extracellular fluid and is rich in fats and proteins. About 0.5 to 1
litre/day of milk is secreted in lactating mothers
Excretion of drugs in milk is a passive process and
is dependent upon pH-partition behaviour, molecular weight, lipid solubility
and degree of ionisation. The pH of milk varies from 6.4 to 7.6 with a mean pH
of 7.0. Free, unionised, lipid soluble drugs diffuse into the mammary alveolar
cells passively. The extent of drug excretion in milk can be determined from
milk/plasma drug concentration ratio (M/P). Since milk is acidic in comparison
to plasma, as in the case of saliva, weakly basic drugs concentrate more in
milk and have M/P ratio greater than 1. The opposite is true for weakly acidic
drugs. It has been shown that for acidic drugs, excretion in milk is inversely
related to the molecular weight and partition coefficient and that for basic drugs,
is inversely related to the degree of ionisation and partition coefficient.
Drugs extensively bound to plasma proteins, e.g. diazepam, are less secreted in
milk. Since milk contains proteins, drugs excreted in milk can bind to it. The
amount of drug excreted in milk is generally less than 1% and the fraction
consumed by the infant is too less to reach therapeutic or toxic levels. But
some potent drugs such as barbiturates, morphine and ergotamine may induce
toxicity in infants. Some examples of toxicity to breast-fed infants owing to
excretion of drug in milk are –
·
Chloramphenicol: Possible bone
marrow suppression.
·
Diazepam: Accumulation and
sedation.
·
Heroin: Prolonged neonatal
dependence.
·
Methadone: Possible withdrawal
syndrome if breast-feeding is stopped suddenly.
·
Propylthiouracil: Suppression of
thyroid function.
·
Tetracycline: Permanent staining
of infant teeth.
Wherever possible, nursing mothers should avoid
drugs. If medication is unavoidable, the infant should be bottle-fed.
5. Skin Excretion
Drugs excreted through the skin via sweat also
follow pH-partition hypothesis. Passive excretion of drugs and their
metabolites through skin is responsible to some extent for the urticaria and
dermatitis and other hypersensitivity reactions. Compounds such as benzoic
acid, salicylic acid, alcohol and antipyrine and heavy metals like lead,
mercury and arsenic are excreted in sweat.
6. Gastrointestinal Excretion
Excretion of drugs into the GIT usually occurs
after parenteral administration when the concentration gradient for passive
diffusion is favourable. The process is reverse of GI absorption of drugs.
Water soluble and ionised form of weakly acidic and basic drugs is excreted in
the GIT, e.g. nicotine and quinine are excreted in stomach. Orally administered
drugs can also be absorbed and excreted in the GIT. Drugs excreted in the GIT
are reabsorbed into the systemic circulation and undergo recycling.
7. Genital Excretion
Reproductive tract and genital secretions may
contain the excreted drugs. Some drugs have been detected in semen.
Drugs can also get excreted via the lachrymal
fluid.
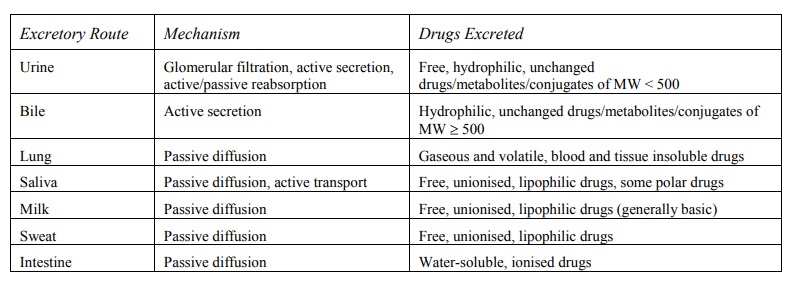
A summary of drugs excreted by various routes is
given in Table 6.4.
TABLE 6.4
Excretion Pathways, Transport Mechanisms and Drugs Excreted
Related Topics
