Properties of colloidal solutions
| Home | | Pharmaceutical Drugs and Dosage | | Pharmaceutical Industrial Management |Chapter: Pharmaceutical Drugs and Dosage: Colloidal dispersions
Properties of colloidal solutions : Kinetic properties, Electrical properties, Colligative properties, Optical properties
Properties of
colloidal solutions
Kinetic properties
Properties
of colloidal systems that arise from the motion of particles with respect to
the dispersion medium are known as kinetic properties. These include Brownian
motion, diffusion, sedimentation, and osmosis.
1. Brownian movement
Brownian
motion results from asymmetry in the force of collisions of mol-ecules of the
dispersion medium on the dispersed phase. This results in ran-dom
dispersed-phase particle motion, called Brownian movement. Since the speed of
motion of the molecules of the dispersion medium increases with temperature,
Brownian motion is a function of temperature. Increase in temperature generally
increases Brownian motion of dispersed-phase par-ticles. The velocity of the
particles also increases with decreasing particle size, which can be attributed
to lower inertia of smaller particles. Similarly, increasing the viscosity of
the medium decreases Brownian movement due to greater resistance to movement of
the dispersed-phase particles.
2. Diffusion
Colloidal
particles are subject to random collisions with other dispersed-phase
particles, usually with a greater force, in addition to the molecules of the
dispersion medium. This leads to the overall movement, called diffu-sion, of
the dispersed-phase particles from a region of high concentration to a region
of low concentration. The rate of diffusion of the dispersed-phase particles is
given by Fick’s first-law equation:

where:
dM is the mass of substance diffusing in
time dt across a cross-sectional area
S
dC/dx
is a concentration gradient
dC over the diffusion distance dx
D is the diffusion
coefficient
The
diffusion coefficient of the dispersed phase, D, is related to the fric-tional coefficient, f, of the particles, which quantitates the resistance to the
movement of particles in the dispersion medium. The diffusion coefficient, D, and the frictional coefficient, f, are inversely related to each other
and are linearly dependent on
temperature, T, as explained by the Einstein’s law of diffusion:
Df = kT
(9.2)
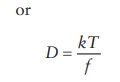
where:
k is the Boltzmann
constant
T is the absolute
temperature
f is the frictional
coefficient
The
Boltzmann constant is a physical constant that relates the average kinetic
energy of particles in a gas with the temperature of the gas and is obtained by
dividing the gas constant, R, by the
Avogadro’s number, N, that is, the
number of molecules per mole of a substance. The Boltzmann constant has the
dimensions of energy over temperature, same as entropy, and is quantitatively
1.38064852(79) × 10−23 J/K. Thus,
k = R/N
The
frictional coefficient is dependent on the size of particles and the viscosity
of the dispersion medium by the equation:
f = 6πηr (9.3)
where:
η is
the viscosity of the medium
r the radius of the
particle
Thus,
diffusion coefficient depends on the viscosity and temperature of the
dispersion medium and the size of the dispersed phase by the equation:
D = kT/6πr (9.4)
This
equation indicates that the diffusion coefficient is inversely propor-tional to
the viscosity of the medium and the radius of the diffusing par-ticles, while
it is directly proportional to the temperature.
Expressing
the Boltzmann constant in terms of the gas constant and the
Avogadro’s
number yields the Stokes–Einstein
equation:
D = RT/6πηrN (9.4)
However,
this equation assumes spherical particles and does not take into account
particle shape effects, which can be important in the case of com-plex
molecules, such as proteins, and linear polymers that can entangle during
movement. In addition, greater the asymmetry or deviation from sphericity,
greater the resistance to flow.
3. Sedimentation
When
stored undisturbed, the dispersed phase tends to separate out from the
dispersion medium and concentrate in one region of the dispersion. When the
dispersed-phase density is higher than that of the dispersion medium, the
dispersed phase accumulates at the bottom, or sediments, and this process is
called sedimentation. This is the case for most aqueous suspensions. When the
dispersed-phase density is lower than that of the dispersion medium, such as in
the case of aqueous emulsions, the dispersed phase accumulates toward the top
of the container, or creams, and this pro-cess is called creaming. Both these
phenomena are governed by the same physics, and for simplicity; this section
will focus on sedimentation.
The
rate of settling of particles, that is, the velocity (v) of sedimentation, is given by the Stokes’ law equation:

where:
ρ
is the density of the particles
ρ0 is the density of the dispersion
medium
η0 is the viscosity of the dispersion
medium
g is the acceleration
due to gravity
In
a centrifugation experiment, g is
replaced by angular acceleration ω2x,
where ω is the angular
velocity and x is the distance of the
particle from the center.
Stokes’
law was derived for dilute dispersions of spherical particles. It does not take
into consideration deviation of particle shape from sphericity and
interparticulate interactions, especially at high dispersed-phase
con-centration. Thus, Stokes’ law may not be quantitatively exactly applicable
to the concentrated dispersions. However, the qualitative, or rank-order,
effects of the factors indicated by the Stokes equation still hold true. For
example, an increase in the mean particle size or in the difference between the
densities of the solid and liquid phases increases the rate of sedimenta-tion.
Using the Stokes equation, creaming of an emulsion or sedimentation of a given
suspension can be reduced by forming smaller particles, increas-ing the
viscosity of continuous phase and/or decreasing the density differ-ence between
two phases.
Electrical properties
Electrical
properties of the dispersed phase refer to the electrostatic charge on the
surface of the particles and its impact on the interaction of the
dis-persed-phase particles with each other and with the dispersion medium.
1. Surface charge
Surface
charge on the dispersed phase plays an important role in the following:
·
Physical stability of colloids. Greater the electrostatic
repulsion among the dispersed-phase particles, greater the physical separation
and uni-form appearance of the colloidal dispersion. However, when settled, the
cake formed may not be easily redispersible. Therefore, a balance of
electrostatic charge on the particles is sought that promotes forma-tion of
uniform dispersion and also allows easy redispersibility on settling.
·
Filtration efficiency of submicron particles, which can be
diminished considerably by particle aggregation or particle affinity for the
filtra-tion membrane.
·
Determining the conformation of macromolecules such as
polymers, polyelectrolytes, and proteins by influencing macromolecule–solvent
interactions and intramolecular interactions within the polymers and
macromolecules.
Most
substances acquire a surface electric charge when brought in con-tact with an
aqueous medium by ionization, ion adsorption, and/or ion dissolution.
1.1 Ionization
Surface
charge arising from ionization on the particles is the function of the pH of
the environment and the pKa
of the particle’s surface functional groups. For example, proteins and peptides
acquire charge through the ionization of surface carboxyl and amino groups to
COO− and NH3+ ions, respectively. The state
and extent of ionization of these groups and the net molecular charge depend on
the pH of the medium and the pKa
of the functional groups, as determined by the Henderson–Hasselbalch equation.
Macromolecules
such as proteins have many ionizable groups. Thus, at pH below its isoelectric
point (PI), the protein molecule bears an overall positively charge, and at pH
above its PI, the protein molecule bears an overall negative charge—even though
there may be domains within the protein structure that would be uncharged or
bear the opposite charge. At the PI of a protein, the total number of positive
charges equals the total number of negative charges in the protein, resulting
in the net charge being zero.
As
an illustration for the amino acid alanine, which has one amino and one
carboxylate group, this phenomenon may be represented as:
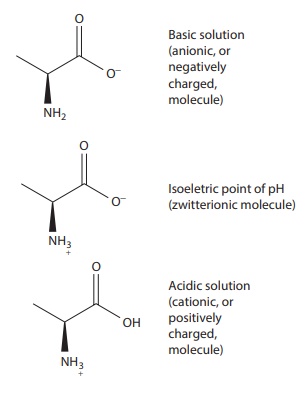
Ionized
molecule has stronger electrostatic, dipole, and hydrogen bond interactions
than unionized functional group or molecule. Thus, ioniza-tion increases
dispersed-phase–aqueous-solvent interactions and generally stabilizes the
dispersion. Addition of salt to solutions of ionized proteins can reduce
protein–solvent interactions and protein solubility. Thus, addi-tion of salt to
precipitate the protein of interest is a common procedure in experimental
sciences. In solutions of multiple proteins, increasing salt concentration can
sequentially precipitate proteins in the increasing order of their aqueous
solubility.
However,
at the isolectric point, proteins with multiple functional groups can
self-associate through interactions of oppositely charged functional groups.
Thus, often, a protein is least soluble at its isoelectric point due to the
attractive interactions between different protein molecules. At the isolectric
point, water-soluble salts such as ammonium sulfate, which par-tially
neutralize surface charges and reduce interparticle attractions, may increase
protein solubility.
1.2 Ion adsorption
Surfaces
that are already charged usually show a tendency to adsorb coun-terions from
solution. For example, a positively charged surface selectively adsorbs
chloride (Cl−) ions from a salt (NaCl) solution. This results in an
excess of the countercharge (i.e., negative charge in the example of Cl−
ions) on the surface of the dispersed phase, compared with the bulk solution. A
second layer of charged coions concentrate above the countercharged surface. In
the aforementioned example, the free Na+ ions in solution form a
second layer over the Cl− ions on the surface. These two layers of
electri-cal charge on a charged surface are together called electrical double layer. Figure 9.1 shows the
electrical double layer on a surface, with the first layer of negatively charged
counterions and the second layer of positively charged
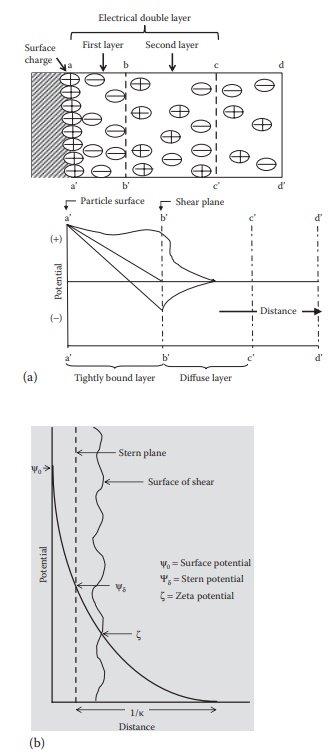
Figure 9.1 Electrical double layer and zeta potential of colloidal particles. (a)
schematic of electrical double layer at the separation between two phases,
showing distri-bution of ions and (b) changes in potential with distance from
particle surface.
A lower net surface charge results on charged surfaces that form electrical
double layer due to unequal adsorption of oppositely charged ions, resulting in
only partial neutralization of particle surface charge.
This
phenomenon can also enable nonpolar surfaces to develop charge by adsorption of
charged solutes from solution. For example, surfactants strongly adsorb by the
hydrophobic effect and determine the surface charge when adsorbed.
1.3 Ion dissolution
Ionic
substances can acquire a surface charge by unequal dissociation of the
oppositely charged ions. For example, in a dispersion of silver iodide
particles with excess [I−] in solution, the dispersed particles
carry a negative charge. This is due to the suppression of dissociation of the
I− ions on the surface of particles by the common-ion effect.
AgI
↔ Ag + + I−
Similarly,
the net charge on AgI particles is positive if excess Ag+ ions are
present in the solution. In this case, therefore, the silver and iodide ions
are referred to as potential-determining
ions, since their concentrations deter-mine the electric potential at the
particle surface.
2. Electrical double layer
As
explained in Section 9.4.2.1.2, the surface
charges of dispersed-phase particles influence the distribution of the nearby
ions in the polar dis-persion medium. Ions with opposite charge (known as counterions) are attracted toward the
surface, and ions with like charges (known as coions) form a second layer on the concentrated layer of
counterions. This leads to the formation of an
electric double layer made up of a neutralizing excess of counterions close
to the charged surface and coions. The electri-cal
double-layer theory explains the distribution of ions with the chang-ing
magnitude of the electric potentials, which occur in the vicinity of the
charged surface.
At
a particular distance from the surface, the concentration of anions and cations
is equal; that is, conditions of electrical neutrality prevail in bulk
solution. The system as a whole is
electrically neutral, even though there are regions of unequal distribution of
anions and cations. This is illustrated in Figure 9.1.
The first layer extends from aa’ to bb’ and is tightly bound to the surface.
This rigid layer attached to the particle surface is called the stern layer. The second layer extends
from bb’ to cc’ and is more diffuse. Hypothetical
planes are defined as boundaries
around the surface of par-ticles that define true hydrated particle size of
electrically double-layered dispersed-phase particles (called stern plane) and the plane that defines
the movement of these particles in solution (called shear plane). The stern plane is at the center of the first layer
of hydrated ions from the surface. The shear plane is the boundary of the first
layer of hydrated ions from the surface.
The
thickness of the electrical double layer is defined by the Debye–Huckel radius or length
parameter, which characterizes the distance from surface at which the particle charge is
completely screened by other charges in solu-tion. This parameter is dependent
on the electrolyte concentration of the aqueous media. The thickness of the
electrical double layer shrinks with increase in electrolyte concentration in
solution.
2.1 Nerst and zeta potentials
Electrothermodynamic
or Nerst potential (E) is defined as the
difference in potential between the
actual surface and the electroneutral region of the solution. This is the
potential at the particle surface (aa’ in Figure 9.1).
However, when the particles are set in motion by electrical forces, such as
electrophoresis, a small layer of solvent with oppositely charged ions moves
concurrently with the particles. The boundary of this layer is termed the shear plane (bb’), since this
distinguishes the moving from the stationary
part of the solvent. The electrical potential at the shear plane bb’ is
known as the electrokinetic or zeta potential, ζ. The ζ potential is defined as the
difference in potential between the surface of the tightly bound layer (shear
plane) and the electroneutral region of the solution.
The
ζ potential, rather
than the Nerst potential, truly governs the degree of repulsion between the
adjacent, similarly charged, dispersed particles. Therefore, measurement and
optimization of ζ potential is needed
for the stability of dispersed systems. The ζ potential can be impacted by all
three mechanisms discussed in the previous section, viz., ionization, ion
dissolu-tion, and ion adsorption. In addition, surfactant molecules that adsorb
by the hydrophobic effect on the surface of the dispersed phase can affect the ζ potential.
2.2 DLVO theory
DLVO
theory is named in honor of Russian physicists B. Derjaguin and L. Landau and
Dutch pioneers in colloid chemistry, E. Verwey and J. Overbreek. These
scientists independently formulated the theories of interaction forces between
colloidal particles in the 1940s to help predict colloidal stability of charged
particles in dispersion. This theory explains the stability of dispersed
colloids in aqueous suspensions on the basis of the balance of two opposite
forces between the dispersed-phase particles: electrostatic force of repulsion
and van der Waals force of attraction.
DLVO
theory of colloidal stability states that the only interactions involved in
determining the stability of colloidal dispersed particles are electric
repulsion (VR) and van der
Waals attraction (VA) and
that these interactions are additive. Therefore, the total potential energy of
interaction (VT) is given
by:
VT =VA +VR (9.7)
Thus,
a stable dispersion is obtained when the repulsive forces dominate, while a
physically unstable dispersion is obtained when attractive forces dominate.
3. Electrophoresis
Electrophoresis
is the movement of charged particles (with the attached ions and the solvent in
the tightly attached first electrical layer) relative to the stationary liquid
dispersion medium, under the influence of an applied electric field. Migration
of particles in an electric field occurs due to the motion of the particle and
its counterion cloud away from the electrode of the same charge and toward the
electrode of the opposite charge.
Electrophoretic
mobility (μ) of a molecule is a
function of its net charge (Q) and
size (radius, r). Thus,
µ =
Q/r
Experimentally,
the electrophoretic mobility is determined as the particle velocity (v) per unit electrical field (E). Thus,
µ
= v/E (9.8)
Hence,
electrophoresis experiments can be used to determine the net sur-face charge on
the particles.
Colligative properties
Colligative
properties are the properties that depend only on the number of nonvolatile molecules in solution, without regard to
their size or molecular weight, or the solute–solute or solute–solvent
interactions.
1. Lowering of vapor pressure
Addition
of a nonvolatile solute to a solvent lowers its vapor pressure, since solute
occupies some of the surface of the solvent. This, therefore, reduces the rate
of evaporation of the solvent.
The
extent of decrease in the vapor pressure with the addition of solute to a
solvent is given by Raoult’s law,
which states that the vapor pressure of an ideal solution is dependent on the
vapor pressure of each individual component, weighted by the mole fraction of
that component in solution. Thus,
PA = XAPA0 (9.9)
where:
PA is the vapor pressure of the colloidal solution
XA is the mole fraction of solute in the solvent
PA0 is the vapor pressure of the pure solvent
Thus,
the reduction of vapor pressure of a solvent is directly proportional to the
concentration of solute in that solvent.
2. Elevation of boiling point
Addition
of a nonvolatile solute leads to the elevation of boiling point due to the
nonvolatile solute displacing the corresponding number of solvent mol-ecules
from the surface and, consequently, reducing the number of solvent molecules
that are able to escape into the vapor phase from the solution. The extent of
increase in the boiling point by the addition of a nonvolatile solute is given
by:
ΔTb
= Kb m (9.10)
where:
Kb is the molal boiling
point elevation constant
ΔTb is the elevation of boiling point
m is the molal amount
of solute in solution
Thus,
the extent of elevation of boiling point is directly proportional to the
concentration of nonvolatile solute in solution. The extent of this effect is
different for each solvent. The value of Kb
for different solvents is available in literature.
3. Depression of freezing point
Addition
of a nonvolatile solute results in reduction in solvent–solvent interactions;
This leads to depression of freezing point of the solvent.
The
extent of decrease in the freezing point is given by:
ΔTf = Kfm (9.11)
where:
Kf is the molal freezing point depression constant
ΔTf is the depression of freezing point
m is the molal amount
of solute in solution
Similar
to the case with the elevation of boiling point, the extent of depression of
freezing point is directly proportional to the concentration of nonvolatile
solute in solution. The extent of this effect is different for each solvent.
The value of Kf for
different solvents is available in literature.
4. Osmotic pressure
Osmosis
involves flow of solvent molecules through a membrane toward its concentration
gradient, which is opposite of the concentration gradient of the solute in
solution. The use of a membrane with a well-defined pore size leads to its
semipermeable nature; that is, only molecules below a certain size or molecular
weight are able to pass through the membrane. Thus, the use of a membrane
through which the colloidal solutes are not able to diffuse, with solutes at
different solution concentration on either side of the membrane, promotes
solvent flow from a solution of low solute concen-tration to a solution of high
solute concentration. This process of solvent flow is called osmosis, and the relative difference in
the pressure of solvent generated by the concentration gradient on either side
of the membrane is called osmotic
pressure, π. The osmotic
pressure is given by:
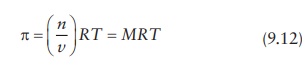
where:
R is the gas constant
T is temperature in
Kelvin
M is the difference in
the molar concentration of solute in solution, which is defined as the number of moles of solute, n, per unit volume of solution, v
Thus,
osmotic pressure of a solution is directly proportional to its solute
concentration and temperature, through their impact on the motion of sol-vent
molecules. The higher the solute concentration and the temperature, the higher
the osmotic pressure.
Optical properties
Colloidal
solutions scatter light, since their particle diameter is within the range of
wavelength of visible light. This phenomenon is known as Tyndall effect. Thus,
light passing through a colloidal solution with particle diameter of ~200 nm leads to scattering,
resulting in turbid or milky appearance. This property is utilized in
quantifying the number of suspended par-ticulates in a liquid or gas colloidal
solution by using a turbidimeter or nephelometer
by calibrating the amount of turbidity at different concentra-tions of a
colloidal solution.
Related Topics
