Resistance To Other Antibiotics
| Home | | Pharmaceutical Microbiology | | Pharmaceutical Microbiology |Chapter: Pharmaceutical Microbiology : Bacterial Resistance To Antibiotics
Chlortetracycline and oxytetracycline were discovered in the late 1940s and studies of representative populations before their widespread use suggests that emergence of resistance is a relatively modern event.
RESISTANCE TO OTHER
ANTIBIOTICS
Resistance To
Tetracycline Antibiotics
Chlortetracycline and oxytetracycline were discovered
in the late 1940s and studies of representative populations before their
widespread use suggests
that emergence of resistance is a relatively modern event. More than 60% of Shigella flexneri
isolates are resistant to tetracycline;
resistant isolates of Salmonella enterica serovar Typhimurium are
becoming more common
and among Gram-positive
species, approximately 90% of MRSA strains and 60% of multiply resistant Strep.
pneumoniae are now tetracycline-resistant. The major mechanisms of resistance are efflux and ribosomal protection. One exception is the tet(X) gene that encodes an enzyme
which modifies
and inactivates the tetracycline molecule, although this does not appear to be clinically significant. The Tet efflux proteins
belong to the major facilitator superfamily
(MFS). These proteins
exchange a proton
for a tetracycline–cation (usually Mg2+) complex,
reducing the intracellular drug concentration and protecting the target ribosomes in the cell. In Gram-negative bacteria, the efflux determinants comprise
divergently oriented efflux and repressor
proteins that share overlapping promoter and operator regions. In the absence of a
tetracycline–Mg2+ complex, the repressor protein
binds and blocks transcription of both genes. Drug binding alters the conformation of the repressor so that it can no longer bind the DNA operator region
and block transcription.
This method of regulation probably
applies to all of the Gram-negative efflux systems including tet(A), tet(C), tet(D), tet(E), tet(G) and tet(H).
No repressor proteins
have been identified in the Gram-positive tet(K)
or tet(L) genes and regulation of plasmid-borne
tetracycline resistance appears
to be by translational attenuation, involving stem-loop mRNA structures and tetracycline-induced unmasking
of the ribosome binding site permitting translation of the efflux protein. Regulation of chromosomal tet(L) expression involves tetracycline-promoted stalling of the ribosomes during translation of early
codons of the
leader peptide, which allows
re-initiation of translation at the ribosome binding site for the structural gene. Ribosomal protection is mediated by cytoplasmic
proteins that inhibit tetracycline and also confer resistance to doxycycline and minocycline. These proteins share homology with the elongation factors
EF-Tu and EF-G, and expression of Tet(M) and Tet(O) proteins
appears to be regulated. A 400-bp region upstream from the coding
region for tet(O) is needed for full expression, but the mechanism(s) has not been
characterized. The widespread emergence of effluxand ribosome
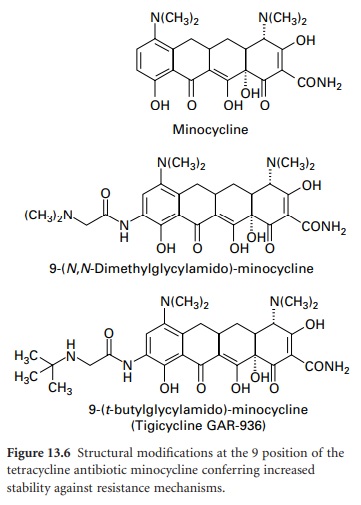
protection-based resistance to firstand second-generation tetracyclines has prompted the development of the 9-glycinyltetracyclines (9glycylcyclines). 9-Amino-acylamido derivatives of
minocycline have similar activity to
earlier compounds; however, when the acyl group is modified to include
an N,N-dialkylamine or 9-t-butyl-glycylamido moiety (Figure
13.6), antimicrobial activity
is retained and the
compounds are active against strains containing tet genes responsible for
resistance by efflux and ribosomal protection.
Resistance To Fluoroquinolone Antibiotics
Fluoroquinolones bind and inhibit
two bacterial topoisomerase enzymes: DNA gyrase (topoisomerase II) which
is required for DNA supercoiling, and
topoisomerase IV which is required for strand separation during cell division. DNA gyrase tends to be the major target in Gram-negative
bacteria, whereas both topoisomerases are inhibited in Gram-positive bacteria. Each topoisomerase is termed a hetero-tetramer, being composed of two copies of two different subunits designated A and B. The A and
B subunits
of DNA gyrases are encoded by gyrA and gyrB,
respectively, whilst topoisomerase IV is encoded by parC and parE (grlA and grlB in
Staph. aureus). Mutations in gyrA, particularly involving substitution of a hydroxyl
group with a bulky hydrophobic group, induce conformational changes such that the fluoroquinolone can no longer bind.
Mutations have also
been detected in the B subunit, but these
are probably less
important. Alterations involving Ser80 and Glu84 of Staph. aureus grlA and Ser79 and Asp83 of Strep. pneumoniae parC
have led to quinolone resistance. Like GyrB, mutations
in ParE leading to resistance are not common.
While changes in GyrA and ParC give
resistance to the
older fluoroquinolones, MIC values
do not always rise above
clinically defined breakpoints for newer agents such as gemi-floxacin and moxifloxacin.
Topoisomerases are located in the cytoplasm and thus fluoroquinolones must cross the cell envelope
to reach their target.
Changes in outer-membrane permeability have been associated with resistance in Gram-negative
bacteria, but permeability does not appear
to be an issue with Gram-positive species.
Efflux, however,
does make a contribution
to resistance, mainly
low level, in both
Gram-positive and Gram-negative bacteria. The NorA mediated efflux system
in Staph. aureus was
characterized in 1990. It is expressed weakly in wild-type strains and
resistance is thought to occur via mutations
leading to increased expression of norA.
NorA is a member of the
MFS and homologues are also
present in Streptococcus
pneumoniae and Bacillus sp.
There is a tendency for it to be
more effective for hydrophilic fluoroquinolones, but there is no strict correlation. Fluoroquinolones are now being used for treating M. avium
and multidrug-resistant
M.
tuberculosis and efflux-mediated
resistance has been identified. A number of efflux pumps have been identified among Gram-negative bacteria, including AcrA in E.coli, which is regulated in part by the multiple-antibiotic resistance
(Mar) operon.
Resistance To
Macrolide, Lincosamide And Streptogramin Antibiotics
Although chemically
distinct, members of the macrolide, lincosamide
and streptogramin (MLS)
group of antibiotics all inhibit bacterial protein synthesis by
binding to a target site on the ribosome. Gram-negative bacteria are intrinsically resistant due to the permeability barrier
of the outer membrane, and three
resistance mechanisms have been described in Gram-positive bacteria. Target modification, involving adenine methylation of domain
V of the 23S ribosomal RNA, is the most common
mechanism. The adenine-N6-methyltransferase, encoded
by the
erm gene, results in resistance to erythromycin and other macrolides (including the azalides), as well as the
lincosamides and group B streptogramins. Streptogramin A-type antibiotics are
unaffected and streptogramin A/B combinations remain effective. Expression of the erm gene may be constitutive or inducible. When expression
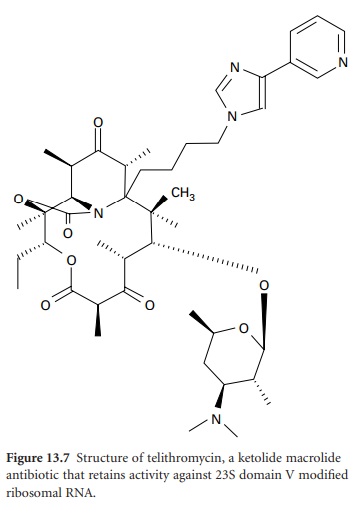
is inducible, resistance is seen only against 14and 15-membered macrolides; lincosamide and streptogramin antibiotics remain active. Telithromycin (Figure 13.7), the first of a new
class of ketolide agents in the
MLS family, does
not induce MLS resistance and
also retains activity against
domain V-modified ribosomes and inhibition of
protein synthesis through strong interaction
with domain II. The second resistance mechanism is efflux. Expression of the mef gene
confers resistance to macrolides only,
whereas msr expression results
in resistance to macrolides
and streptogramins. Efflux-mediated resistance
of Staph. aureus to
streptogramin A antibiotics is also conferred by vga and
vgaB gene products. A third
resistance mechanism, involving ribosomal mutation, has been reported in a small number of clinical isolates
of Strep. pneumoniae.
Resistance To
Chloramphenicol
Chloramphenicol inhibits
protein synthesis by binding
the 50S ribosomal subunit and preventing the peptidyltransferase
step. Decreased outer-membrane permeability and active
efflux have been
identified in Gram-negative bacteria; however,
the major resistance mechanism is
drug inactivation by
chloramphenicol acetyltransferase. This occurs in both Gram-positive and Gram-negative
species, but the cat genes, typically found on plasmids, share little
homology.
Resistance To The
Oxazolidinone Antibiotics
Linezolid is the
first of a new class
of oxazolidinone antimicrobials with a novel target
in protein synthesis. Linezolid does not
interfere with translation initiation at the stage
of mRNA binding
or formation of 30S preinitiation complexes; rather, it involves
binding the 50S rRNA.
Its affinity for 50S rRNA from Gram-positive bacteria is twice that for the corresponding molecule in Gram negative bacteria and as such linezolid has been approved for treating various
Gram-positive infections, including MRSA. Resistance is appearing, although
rare at present. Mutation in the central loop of domain
V of the component 23S rRNA subunit
appears to be the main
mechanism,
including a G2576T mutation in three isolates of linezolid-resistant MRSA.
Resistance To
Trimethoprim
Trimethoprim
competitively inhibits dihydrofolate reductase (DHFR) and
resistance can be caused by overproduction of host DHFR, mutation in
the structural gene for DHFR
and acquisition of the dfr gene
encoding a resistant form. There are
at least 15 DHFR enzyme
types based on sequence
homology and acquisition of dfr genes
encoding alternative DHFR of type
I, II or V is the most common mechanism of trimethoprim resistance
among the Enterobacteriaceae.
Resistance To Mupirocin
Nasal carriage of MRSA
strains has been identified as an important target for infection control
protocols aimed at reducing spread and acquisition. Mupirocin (pseudomonic acid
A) is an effective topical antimicrobial used in MRSA eradication. It is an analogue
of isoleucine that competitively binds isoleucyl-tRNA synthetase (IRS) and
inhibits protein synthesis. Low-level resistance (MIC 4–256 μg/ml) is usually
due to mutation of the host IRS, whereas high-level resistance (MIC >512
μg/ml) is due to acquisition of a distinct IRS that is less sensitive to inhibition.
The mupA gene, typically carried on transferable plasmids, is found in Staph.
aureus and co-agulasenegative staphylococci, and encodes an IRS with only 30%
homology to the mupirocin-sensitive form.
Resistance To Peptide Antibiotics—Polymyxin
Many peptide
antibiotics have been described and can be broadly classified as non-ribosomally synthesized peptides; they include the polymyxins, bacitracins and gramicidins as well as the glycopeptides (section
5) and the
ribosomally synthesized peptides such as the
antimicrobial peptides of the innate immune system. Polymyxins and other
cationic antimicrobial peptides have a self-promoted uptake across the cell envelope and perturb the cytoplasmic membrane barrier. Addition of a 4-amino-4-deoxy-l-arabinose (l-Ara4N) moiety to the phosphate groups on the lipid A component of Gram-negative lipopolysaccharide has been implicated in resistance to polymyxin. Details of the pathway for l-Ara4N biosynthesis from UDP glucuronic acid, encoded by the pmr operon, are emerging.
Resistance To Antimycobacterial Therapy
The nature of mycobacterial
infections, particularly tuberculosis, means that chemotherapy differs from
other infections. Organisms tend to grow slowly (long generation time) in a near
dormant state with
little metabolic activity. Hence, a number
of the conventional antimicrobial targets
are not suitable. Isoniazid is bactericidal, reducing the count of aerobically growing
organisms. Pyrazinamide is active only
at low pH, making it well
suited to killing organisms
within necrotic foci early in infection, but less useful later on when these foci have reduced in number. Rifampicin targets slow-growing organisms. Resistance
mechanisms have now been described and multiple resistance poses a serious threat to health. Current
treatment regimens result in a high
cure rate and the combination of agents makes
it highly unlikely that there
will be a spontaneous resistant
isolate to all
the components. Problems
most commonly occur in patients who receive
inadequate therapy, which
provides a serious selection advantage. Resistance can occur
to single agents and subsequently to multiple agents. Resistance to rifampicin arises
from mutation in the β subunit of RNA polymerase encoded
by rpoB and resistant isolates show decreased growth
rates. Modification of the catalase gene katG results in resistance to isoniazid,
mainly by reduced or absent catalase
activity. Catalase activity is absolutely required to convert isoniazid to the active hydrazine derivative. Interestingly,
animal model studies suggest that M.
tuberculosis strains in which the
katG gene is inactivated are attenuated compared
with wild-type
strains. Low-level rifampicin resistance can be obtained by point
mutations in inhA leading to its overexpression. Pyrazinamide is a prodrug requiring
pyrazinamidase to
produce the active pyrazinoic acid. Most
cases of resistance are due to mutations
in the pyrazinamidase gene (pncA), but gene inactivation by the insertion sequence IS6110 has
been reported. Streptomycin resistance can arise through
mutations in rrs and
rpsL which affect streptomycin binding. However, these account for only half of the resistant isolates, so further resistance mechanisms await
definition. Ethambutol resistance has been noted in M. tuberculosis and
other species such as M. smegmatis. Ethambutol inhibits the polymerization of arabinan in the
arabinogalactan and lipo-arabinomannan of the mycobacterial cell wall and one of its likely targets
is the family of arabinosyl-transferases encoded by the emb locus. Missense mutations in the embB gene in this locus
confer resistance to ethambutol.
Related Topics
