Stability
| Home | | Pharmaceutical Drugs and Dosage | | Pharmaceutical Industrial Management |Chapter: Pharmaceutical Drugs and Dosage: Pharmaceutical solutions
Stability: Physical stability, Chemical stability, Microbial stability
Stability
Physical stability
A
solution consists of drug substance solubilized in a vehicle commonly with the
aid of pH control, surfactant(s), or cosolvent(s). Physical or chemi-cal
changes during storage, such as decrease in the storage temperature, microbial
growth resulting in pH change and cosolvent evaporation or loss by selective
adsorption, can lead to supersaturation of the drug in the vehi-cle.
Supersaturated solutions can form crystal nuclei of the drug when the
supersaturated drug concentration reaches above the threshold for nucle-ation (Figure 18.1). The crystal nuclei tend to grow slowly
(crystallization)
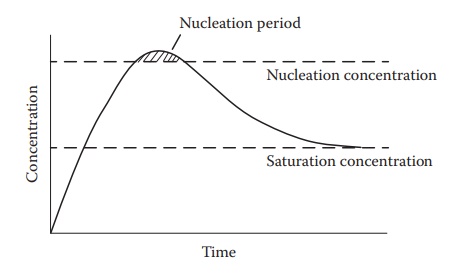
Figure 18.1 Time dependence of concentration required for monodispersity. This
figure represents supersaturation region of drug solubility between saturation
and the concentration that leads to nucleation. (From Narang, A.S. et al., Int.
J. Pharm. 345, 9–25, 2007. With Permission.)
In
addition, sudden changes in temperature, such as freezing, can result in
instantaneous precipitation of the
drug in the form of small, amor-phous particles. Formulating a drug solution
much below its saturation concentration is preferred to avoid physical
instability by precipitation or crystallization.
Chemical stability
Solution
dosage form presents an environment with high molecular mobil-ity of reacting
species, resulting in higher degradation liability than other dosage forms,
such as tablets. Common modes of drug degradation in solution include
hydrolysis and oxidation. Drug degradation pathways and stabilization
strategies are discussed in Chapter 7.
Degradation
of drug in the dosage form leads to decrease in drug potency and formation of
impurities. Depending on the therapeutic window and dose of the drug, and the
toxicological nature and quantity of impurities formed, the national compendia
such as the United States Pharmacopeia (USP) and the international bodies such
as the International Council on Harmonization (ICH) recommend maximum limits on
the permissible impurities. These limits are identified in terms of reporting,
identification, or qualification thresholds—requiring the sponsor of the new
drug applica-tion (NDA) to report, identify, or provide toxicological safety
data on the given impurity to identify and justify a maximum permissible
concentra-tion. In addition, impurities that are suspected to be genotoxic are
rigor-ously controlled.
In
addition to chemical stability of the drug, adequate potency of other additives
critical to the stability and performance of the dosage form, such as
antimicrobial agents and antioxidants, must be demonstrated through-out a
product’s shelf life.
Microbial stability
Pharmaceutical
aqueous solutions generally contain organic com-pounds, including
carbohydrates, thus providing a suitable growth environment for bacteria and
other microbes. Except in the case of broad-spectrum antibiotics or
self-preserving solutions, such as syrups, antimicrobial preservatives are
frequently required in solution formu-lations. Methylparaben, propylparaben,
and sodium benzoate are the commonly used antimicrobial agents. Methylparaben
and propylpara-ben are commonly used in 9:1 w/w ratio at combination at 0.2%
w/v total concentration.
Related Topics
