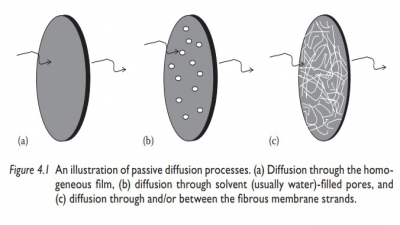
Absorption
| Home | | Pharmaceutical Drugs and Dosage | | Pharmaceutical Industrial Management |Chapter: Pharmaceutical Drugs and Dosage: Biopharmaceutical considerations
Bioavailability is the fraction of an ingested dose of a drug that is absorbed into the systemic circulation, compared with the same dose of the com-pound injected intravenously, which is directly injected into the systemic circulation.
Absorption
Bioavailability is the fraction of
an ingested dose of a drug that is absorbed
into the systemic circulation, compared with the same dose of the com-pound
injected intravenously, which is directly injected into the systemic
circulation. Bioavailability of a drug is determined during new product
development.
Bioequivalence, on the other hand,
is a comparison of relative bio-availability of two dosage forms in terms of
the rate and extent of the drug levels achieved in the systemic circulation and
the maximum drug concentration reached. Generic drugs are required to satisfy
statistical cri-teria of bioequivalence to the branded version before they can
be considered equivalent.
In
the case of oral dosage forms, drug bioavailability depends on the rate and
extent of drug absorption from the GI tract. Drug absorption from the gut
depends on many factors, such as the drug’s solubility and intrinsic
dis-solution rate in the GI fluids, which are influenced by GI pH and motility,
and the drug’s particle size and surface area. Thus, an interplay of
physico-chemical properties of the drug and physiological properties of the GI
tract determines the outcome of factors that determine drug absorption.
Drug
absorption is affected not only by the properties of drug and its dosage forms
but also by the nature of the biological membranes. Drugs pass through living
membranes by the following processes (Figure 4.4):
1.
Passive diffusion
a.
Simple diffusion
b.
Facilitated diffusion
i.
Channel-mediated transport
ii.
Carrier-mediated transport
2.
Active transport
Passive
diffusion can also be classified as paracellular or transcellular, depending on
the route of drug absorption across the epithelial cell barrier. The surface
lining of the GI tract consists of epithelial cells attached to each other by
tight junctions formed through their membranes. Drug transport across the tight
junctions between cells is known as paracellular
transport. It involves both diffusion and the convective flow of water
accompany-ing water-soluble drug molecules. Drug transport by absorption into
the
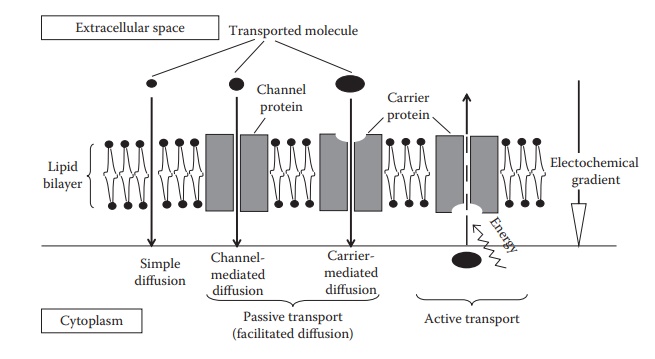
Figure 4.4 An illustration of the main
transport processes across cellular membranes.
epithelial
cell from the gut’s lumen side, followed by release of the drug molecule from
the epithelial membrane on the other side of the epithelial cell into the
systemic circulation, is known as transcellular
transport.
Passive transport
Passive
transport can be divided into simple diffusion, carrier-mediated dif-fusion,
and channel-mediated diffusion (Figure 4.4).
Simple diffusion
Biological
membranes are lipoidal in nature; that is, they are made of lipid bilayers,
with hydrophobic tails in the center and hydrophilic heads fac-ing the aqueous
environment on either side. Therefore, hydrophobic lipid-soluble drugs of low
molecular weight can pass through membranes by simple diffusion. Passive
transport by simple diffusion is driven by differ-ences in drug concentration
on the two sides of the membrane. In intestinal absorption, for example, the drug
travels by passive transport from a region of high concentration in the GI
tract to a region of low concentration in the systemic circulation. Given the
instantaneous dilution of the absorbed drug once it reaches the bloodstream,
sink conditions are essentially maintained at all times.
Carrier-mediated transport
Carrier-mediated
transport is a passive diffusion process that involves facil-itation or
increase of diffusion rate by the involvement of a carrier protein embedded in
the biological membrane. It differs from active transport in that the drug
moves along a concentration gradient (i.e., from a region of high concentration
to the one of low concentration) and that this system does not require energy
input; that is, the carrier does not use energy, such as adenosine triphosphate
(ATP), to transport the drug. Carrier-mediated transport is saturable,
structurally selective for the drug, and shows com-petition kinetics for drugs
of similar structures. Carrier-mediated transport does not require the substrate
to be lipophilic: both hydrophilic and lipo-philic solutes can be transported
in this manner.
Amino
acid transporters, oligopeptide transporters, glucose transport-ers, lactic
acid transporters, phosphate transporters, bile acid transporters, and other
transporters facilitate drug transport across the GI tract, espe-cially the
small intestine. Transporters are
specific proteins in the biological membranes that transport the molecules
(e.g., glucose) across the mem-brane. Transporters bind to the molecule,
transport the molecule across the membrane, and then release it on the other
side. The transporter remains unchanged after the process.
Channel-mediated transport
A
fraction of the cell membrane is composed of aqueous-filled pores or channels,
which are continuous across the membrane. These pores offer a pathway parallel
to the diffusion pathway through the lipid bilayer. Channel-mediated transport
(also known as port or convective transport) plays an important role in the
transport of ions and charged drugs, espe-cially in the case of renal excretion
and hepatic uptake of drugs. Certain transport proteins may form an open
channel across the lipid membrane of the cell. Small molecules, including
drugs, move more rapidly through the channel by diffusion than by simple
diffusion across the membrane due to facilitation by the solvent and if their
diffusion rate in the solvent is higher than in the lipoidal membrane.
Fick’s laws of diffusion in drug absorption
Transport
of a drug by diffusion across a membrane such as the GI mucosa is represented
by Fick’s law equation:

where:
M is the amount of
drug in the gut compartment at time t
Dm is the diffusion coefficient or diffusivity of the drug in
intestinal membrane
Smembrane is the surface area of the membrane
Kmembrane/intestinal ⋅fluid is the partition coefficient of the drug between the membrane and the aqueous intestinal
fluid
hmembrane is the membrane’s thickness
Cgut is the drug concentration in the gut or intestinal
compartment
Cplasma is the drug concentration in plasma compartment
Since
the absorbed drug is instantaneously diluted and rapidly removed from the
absorption site by the systemic circulation, Cplasma →
0. Therefore, Equation 4.26 becomes:

The
left-hand side of Equation 4.27 can be converted
into concentration units, since:
Cgut = M/V
On
the right-hand side of the equation, the diffusion coefficient, membrane area,
partition coefficient, and membrane thickness can be combined to yield a
permeability coefficient.
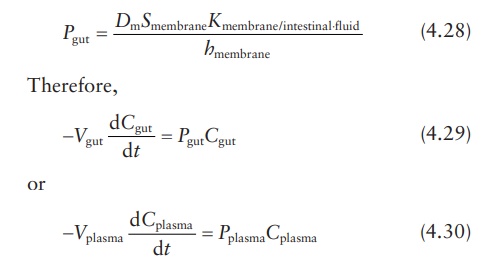
where
Cgut and Pgut are the concentration
and permeability coefficient, respectively, for drug passage from intestine to
plasma. Similarly, Cplasma
and Pplasma are the
concentration and permeability coefficient, respectively, for the reverse
passage of drug from plasma to intestine. These equations demonstrate that the
ratio of absorption rates in the intestine-to-plasma and the
plasma-to-intestine directions depends on the ratio of permeability
coefficients, drug concentrations, and volumes of drug distribution.
Active transport
Active
transport involves the use of transmembrane proteins that require the use of
cellular energy (usually ATP) to actively pump substances into or out of the
cell. In active transport, the molecules usually move from regions of low
concentration to those of high concentration. The most well-known active
transport system is the sodium–potassium–ATPase pump (Na+/K+
ATPase), which maintains an imbalance of sodium and potassium ions inside and
outside the membrane, respectively, for neuronal signal transmission. The Na+/K+
pump is an antiport; it transports K+ into the cell and Na+
out of the cell at the same time, with no expenditure of ATP. Other active
trans-port systems include the sodium–hydrogen ion pump of the GI tract, which
maintains gastric acidity while absorbing sodium ions, and the calcium ion
pump, which helps maintain a low concentration of calcium in the cytosol.
Related Topics