Chapter Summary, Questions Answers - Cholesterol, Lipoprotein, and Steroid Metabolism
| Home | | Biochemistry |Chapter: Biochemistry : Cholesterol, Lipoprotein, and Steroid Metabolism
Cholesterol is a hydrophobic compound, with a single hydroxyl group located at carbon 3 of the A ring, to which a fatty acid can be attached, producing an even more hydrophobic cholesteryl ester.
CHAPTER SUMMARY
Cholesterol is a
hydrophobic compound, with a single hydroxyl group located at carbon 3 of the A
ring, to which a fatty acid can be attached, producing an even more hydrophobic
cholesteryl ester. Cholesterol is synthesized by virtually all human tissues,
although primarily by liver, intestine, adrenal cortex, and reproductive
tissues (Figure 18.29). All the carbon atoms in cholesterol are provided by
acetyl coenzyme A (CoA), and nicotinamide adenine dinucleotide phosphate
provides the reducing equivalents. The pathway is driven by hydrolysis of the
high-energy thioester bond of acetyl CoA and the terminal phosphate bond of
adenosine triphosphate. Cholesterol is synthesized in the cytoplasm. The
rate-limiting and regulated step in cholesterol synthesis is catalyzed by the
smooth endoplasmic reticulum–membrane protein, hydroxymethylglutaryl coenzyme A
(HMG CoA) reductase, which produces mevalonate from HMG CoA. The enzyme is
regulated by a number of mechanisms: 1) Expression of the gene for the reductase is
activated when cholesterol levels are low, via the transcription factor, sterol
regulatory element–binding protein-2 (SREBP-2), bound to a sterol regulatory element
(SRE), resulting in increased enzyme and, therefore, more cholesterol,
synthesis; 2) degradation of the reductase is accelerated when cholesterol
levels are high; 3) reductase activity is controlled by adenosine monophosphate
(AMP)–activated protein kinase ([AMPK], which phosphorylates and inactivates
the reductase) and an insulin-activated protein phosphatase (which
dephosphorylates and activates it); and 4) expression of the gene for the
reductase is upregulated by insulin and downregulated by glucagon. Statins are
competitive inhibitors of HMG CoA reductase. These drugs are used to decrease
plasma cholesterol in patients with hypercholesterolemia. The ring structure of
cholesterol cannot be degraded in humans.
Cholesterol can be
eliminated from the body either by conversion to bile salts or b y secretion
into the bile. Bile salts and phosphatidylcholine are quantitatively the most
important organic components of bile. The rate-limiting step in bile acid
synthesis is catalyzed by cholesterol-7-gα-hydroxylase, which is inhibited by
bile acids. Before the bile acids leave the liver, they are conjugated to a
molecule of either glycine or taurine, producing the conjugated bile salts glycocholic
or taurocholic acid and glycochenodeoxycholic or taurochenodeoxycholic acid.
Bile salts (deprotonated) are more amphipathic than bile acids (protonated)
and, therefore, are more effective emulsifiers of dietary fat. In the
intestine, bacteria can remove the glycine and taurine and can remove a
hydroxyl group from the steroid nucleus, producing the secondary bile salts,
deoxycholic and lithocholic acids. More than 95% of the bile salts are
efficiently reabsorbed in the intestinal ileum by a sodium–bile salt
cotransporter, returned to the blood, and carried by albumin back to the liver
where they are taken up by the hepatic form of the cotransporter and reused
(enterohepatic circulation, which bile acid sequestrants reduce). If more
cholesterol enters the bile than can be solubilized by the available bile salts
and phosphatidylcholine, cholesterol gallstone disease (cholelithiasis) can
occur.
The plasma lipoproteins
include chylomicrons, very-low-density lipoproteins (VLDLs) , low-density
lipoproteins (LDLs), and high-density lipoproteins (HDLs). They function to
keep lipids (primarily triacyl-glycerol [TAG] and cholesteryl esters) soluble
as they transport them between tissues. Lipoproteins are composed of a neutral
lipid (TAG, cholesteryl esters, or both) core surrounded by a shell of
amphipathic apolipoproteins, phospholipid, and unesterified cholesterol.
Chylomicrons are assembled in intestinal mucosal cells from dietary lipids
(primarily TAG). Each nascent chylomicron particle has one molecule of
apolipoprotein (apo) B-48. They are released from the cells into the lymphatic
system and travel to the blood, where they receive apo C-II and apo E from
HDLs. Apo C-II activates endothelial lipoprotein lipase (LPL), which degrades
the TAG in chylomicrons to fatty acids and glycerol. The fatty acids that are
released are stored (in the adipose) or used for energy (by the muscle). The
glycerol is metabolized by the liver. Patients with a deficiency of LPL or apo
C-II show a dramatic accumulation of chylomicrons in the plasma (type I
hyperlipoproteinemia, or familial LPL deficiency) even if fasted. After most of
the TAG is removed, apo C-II is returned to the HDL, and the chylomicron
remnant, carrying most of the dietary cholesterol, binds to a receptor on the
liver that recognizes apo E. The particle is endocytosed, and its contents
degraded by lysosomal enzymes. Defective uptake of these remnants causes type
III hyperlipoproteinemia. Nascent VLDLs are produced in the liver and are
composed predominantly of TAG. They contain a single molecule of apo B-100.
Like nascent chylomicrons, VLDLs receive apo C-II and apo E from HDL in the
plasma. The function of VLDL is to carry hepatic TAG to the peripheral tissues
where LPL degrades the lipid. Additionally, the VLDL particle receives
cholesteryl esters from HDL in exchange for TAG. This process is accomplished
by cholesteryl ester transfer protein (CETP). Eventually, VLDL in the plasma is
converted to LDL, a much smaller, denser particle. Apo C-II and apo E are
returned to HDL, but the LDL retains apo B-100, which is recognized by receptors
o n peripheral tissues and the liver. LDLs undergo receptor-mediated
endocytosis, and their contents are degraded in the lysosomes. A deficiency of
functional LDL receptors causes type II hyperlipoproteinemia (familial
hypercholesterolemia). The endocytosed cholesterol decreases synthesis of HMG
CoA reductase (and of LDL receptors) through prevention of SREBP-2 binding to
the SRE. Some of it can also be esterified by acyl CoA:cholesterol
acyltransferase (ACAT) and stored. HDL are created by lipidation of apo A-1
synthesized in the liver and intestine. They have a number of functions,
including: 1) serving as a circulating reservoir of apo C-II and apo E for
chylomicrons and VLDL; 2) removing unesterified cholesterol from from
peripheral tissues via ABCA1and esterifying it using lecithin:cholesterol acyl
transferase(LCAT), a liver-synthesized plasma enzyme that is activated by apo
A-1; and 3) delivering these cholesteryl esters to the liver (reverse
cholesterol transport) for uptake via scavenger receptor-B1(SR-B1).
Cholesterol is the precursor of all classes of steroid hormones, which include glucocorticoids, mineralocorticoids, and the sex hormones (androgens, estrogens, and progestins) . Synthesis, using primarily cytochrome P450 mixed-function oxidases, occurs in the adrenal cortex (cortisol, aldosterone, and androgens), ovaries and placenta (estrogens and progestins), and testes (testosterone). The initial and rate-limiting step is the conversion of cholesterol to pregnenolone by the side-chain cleavage enzyme P450scc. Deficiencies in synthesis Iead to congenital adrenal hyperplasia. Each steroid hormone diffuses across the plasma membrane of its target cell and binds to a specific cytosolic or nuclear receptor. These receptor–ligand complexes accumulate in the nucleus, dimerize, and bind to specific regulatory DNA sequences (hormone-response elements) in association with coactivator proteins, thereby causing promoter activation and increased transcription of targeted genes. In association with corepressors, transcription is decreased.
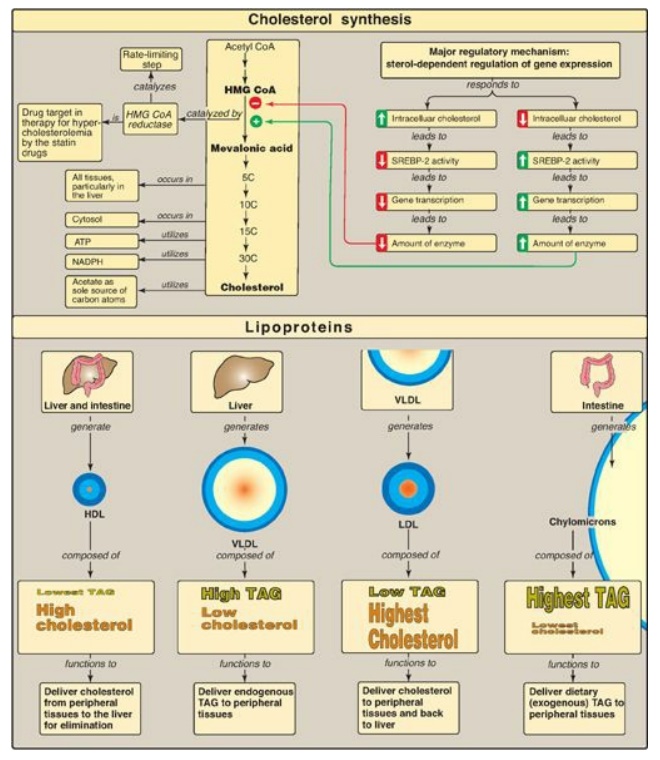
Figure 18.29 Concept map for cholesterol and the lipoproteins. HMG CoA = hydroxymethylglutaryl coenzyme A; SREBP = sterol regulatory element-binding protein; HDL = high-density lipoprotein; VLDL = very-low-density lipoprotein; LDL = lowdensity lipoprotein; TAG = triacylglycerol; NADP(H) = nicotinamide adenine dinucleotide phosphate.
Study Questions
Choose the ONE best answer.
18.1 Mice were genetically engineered to contain
hydroxymethylglutaryl coenzyme A reductase in which serine 871, a
phosphorylation site, was replaced by alanine. Which of the following
statements concerning the modified form of the enzyme is most likely to be
correct?
A. The enzyme is nonresponsive to adenosine
triphosphate depletion.
B. The enzyme is
nonresponsive to statin drugs.
C. The enzyme is
nonresponsive to the sterol response element–sterol response element–binding
protein system.
D. The enzyme is unable
to be degraded by the ubiquitin–proteasome system.
Correct answer = A. The reductase is regulated by covalent phosphorylation and dephosphorylation. Depletion of adenosine triphosphate results in a rise in adenosine monophosphate (AMP), which activates AMP kinase (AMPK), thereby phosphorylating and inactivating the enzyme. In the absence of the serine, a common phosphorylation site, the enzyme cannot be phosphorylated by AMPK. The enzyme is also regulated physiologically through changes in transcription and degradation and pharmacologically by statin drugs (competitive inhibitors), but none of these depends on serine phosphorylation.
18.2 Calculate the amount of cholesterol in the
low-density lipoproteins in an individual whose fasting blood gave the
following lipid-panel test results: total cholesterol = 300 mg/dl, high-density
lipoprotein cholesterol = 25 mg/dl, triglycerides = 150 mg/dl.
A. 55 mg/dl
B. 95 mg/dl
C. 125 mg/dl
D. 245 mg/dl
Correct answer = D. The total cholesterol in the blood
of a fasted individual is equal to the sum of the cholesterol in low-density
lipoproteins plus the cholesterol in high-density lipoproteins plus the
cholesterol in very-low-density lipoproteins (VLDLs). This last term is
calculated by dividing the triacylglycerol value by 5 because cholesterol
accounts for about 1/5 of the volume of VLDL in fasted blood.
For Questions 18.3 and
18.4:
A young girl with a history of severe abdominal
pain was taken to her local hospital at 5 a.m. in severe distress. Blood was
drawn, and the plasma appeared milky, with the triacylglycerol level in excess
of 2,000 mg/dl (normal = 4–150 mg/dl). The patient was placed on a diet
extremely limited in fat but supplemented with medium-chain triglycerides.
Correct answer = A. The milky appearance of her blood
was a result of triacylglycerol-rich chylomicrons. Because 5 a.m. is presumably
several hours after her evening meal, the patient must have difficulty
degrading these lipoprotein particles. Intermediate-, low-, and high-density
lipoproteins contain primarily cholesteryl esters, and, if one or more of these
particles was elevated, it would cause hypercholesterolemia. Very-low-density
lipoproteins do not cause the described “milky appearance” in plasma.
18.3 Which of the following lipoprotein particles
are most likely responsible for the appearance of the patient’s plasma?
A. Chylomicrons
B. High-density
lipoproteins
C. Intermediate-density lipoproteins
D. Low-density
lipoproteins
E. Very-low-density
lipoproteins
Correct answer = C. The triacylglycerol (TAG) in
chylomicrons is degraded by endothelial lipoprotein lipase, which requires apo
C-II as a coenzyme. Deficiency of the enzyme or coenzyme results in decreased
ability to degrade chylomicrons to their remnants, which get cleared by the
liver. Apo A-I is the coenzyme for lecithin:cholesterol acyltransferase; apo
B-48 is the ligand for the hepatic receptor that binds chylomicron remnants;
cholesteryl ester transfer protein catalyzes the cholesteryl ester–TAG exchange
between high-density and very-low-density lipoproteins (VLDLs); and microsomal
triglyceride transfer protein is involved in the formation, not degradation, of
chylomicrons (and VLDLs).
18.4 Which one of the following proteins is most
likely to be deficient in this patient?
A. Apo A-I
B. Apo B-48
C. Apo C-II
D. Cholesteryl ester
transfer protein
E. Microsomal
triglyceride transfer protein
18.5 Complete the table
below for an individual with classic 21-α-hydroxylase deficiency relative to a
normal individual.
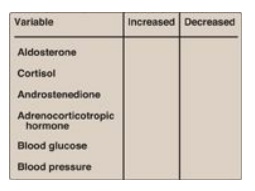
How might the results
be changed if this individual were deficient in 17-α-hydroxylase, rather than
21-α-hydroxylase?
21-α-Hydroxylase
deficiency causes mineralocorticoids (aldosterone) and glucocorticoids
(cortisol) to be virtually absent. Because aldosterone increases blood
pressure, and cortisol increases blood glucose, their deficiencies result in a
decrease in blood pressure and blood glucose, respectively. Cortisol normally
feeds back to inhibit adrenocorticotropic hormone (ACTH) release by the
pituitary, and, so, its absence results in an elevation in ACTH. The loss of
21-α-hydroxylase pushes progesterone and pregnenolone to androgen synthesis,
therefore, causes androstenedione levels to rise.
With 17-α-hydroxylase
deficiency, sex hormone synthesis would be inhibited. Mineralocorticoid
production would be increased, leading to hypertension.
Related Topics
