Metabolism of Ammonia
| Home | | Biochemistry |Chapter: Biochemistry : Amino Acids: Disposal of Nitrogen
Ammonia is produced by all tissues during the metabolism of a variety of compounds, and it is disposed of primarily by formation of urea in the liver.
METABOLISM OF AMMONIA
Ammonia is produced by
all tissues during the metabolism of a variety of compounds, and it is disposed
of primarily by formation of urea in the liver. However, the level of ammonia
in the blood must be kept very low, because even slightly elevated
concentrations (hyperammonemia) are toxic to the central nervous system (CNS).
Therefore, there must be a metabolic mechanism by which nitrogen is moved from
peripheral tissues to the liver for ultimate disposal as urea, at the same time
maintaining low levels of circulating ammonia.
A. Sources of ammonia
Amino acids are quantitatively the most important source of ammonia because most Western diets are high in protein and provide excess amino acids, which travel to the liver and undergo transdeamination (that is, the linking of aminotransferase and glutamate dehydrogenase reactions), producing ammonia. [Note: Liver catabolizes straight-chain amino acids, primarily.] However, substantial amounts of ammonia can be obtained from other sources.
1. From glutamine: An important source of plasma
glutamine is from the catabolism of branched-chain amino acids in skeletal
muscle. This glutamine is taken up by cells of the intestine, the liver, and
the kidney. The liver and kidneys generate ammonia from glutamine by the
actions of glutaminase (Figure 19.17) and glutamate dehydrogenase. In the
kidneys, most of this ammonia is excreted into the urine as NH4+,
which provides an important mechanism for maintaining the body’s acid–base
balance through the excretion of protons. In the liver, the ammonia is
detoxified to urea and excreted. [Note: α-Ketoglutarate, the second product of
glutamate dehydrogenase, is a glucogenic precursor in liver and kidney.]
Ammonia is also generated by intestinal glutaminase. The intestinal mucosal
cells obtain glutamine either from the blood or from digestion of dietary
protein. [Note: Intestinal glutamine metabolism also produces alanine, which is
used by the liver for gluconeogenesis, and citrulline, which is used by the kidneys
to synthesize arginine.]
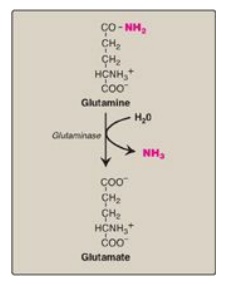
Figure 19.17 Hydrolysis of glutamine to form ammonia (NH3).
2. From bacterial action in the intestine: Ammonia is formed from urea by the
action of bacterial urease in the lumen of the intestine. This ammonia is
absorbed from the intestine by way of the portal vein, and virtually all is
removed by the liver via conversion to urea.
3. From amines: Amines obtained from the diet and monoamines that
serve as hormones or neurotransmitters give rise to ammonia by the action of
monoamine oxidase.
4. From purines and pyrimidines: In the catabolism of purines and
pyrimidines, amino groups attached to the ring atoms are released as ammonia
(see Figure 22.15).
B. Transport of ammonia in the circulation
Although ammonia is
constantly produced in the tissues, it is present at very low levels in blood.
This is due both to the rapid removal of blood ammonia by the liver and to the
fact that several tissues, particularly muscle, release amino acid nitrogen in
the form of glutamine or alanine, rather than as free ammonia (see Figure
19.13).
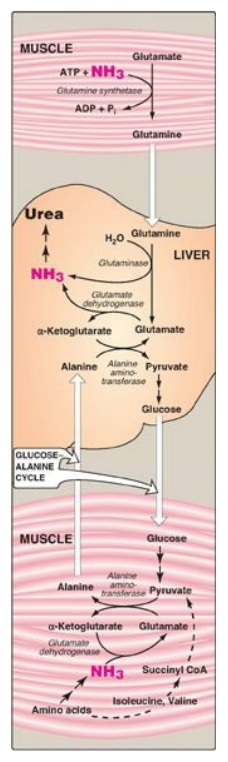
Figure 19.13 Transport of ammonia (NH3) from muscle to the liver. ADP = adenosine diphosphate; Pi = inorganic phosphate; CoA = coenzyme A.
1. Urea: Formation of urea in the liver is quantitatively
the most important disposal route for ammonia. Urea travels in the blood from
the liver to the kidneys, where it passes into the glomerular filtrate.
U2. Glutamine: This amide of glutamate provides a nontoxic
storage and transport form of ammonia (Figure 19.18). The ATP-requiring
formation of glutamine from glutamate and ammonia by glutamine synthetase
occurs primarily in skeletal muscle and liver but is also important in the CNS,
where it is the major mechanism for the removal of ammonia in the brain.
Glutamine is found in plasma at concentrations higher than other amino acids, a
finding consistent with its transport function. [Note: The liver keeps blood
ammonia levels low through glutaminase and the urea cycle in periportal (close
to inflow of blood) hepatocytes and via glutamine synthetase as an ammonia
“scavenger” in the perivenous hepatocytes.] The metabolism of ammonia is
summarized in Figure 19.19.
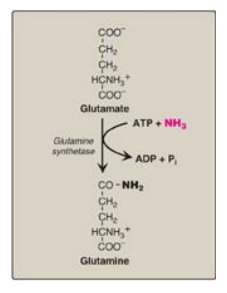
Figure 19.18 Synthesis of glutamine. ADP = adenosine diphosphate; Pi = inorganic phosphate.
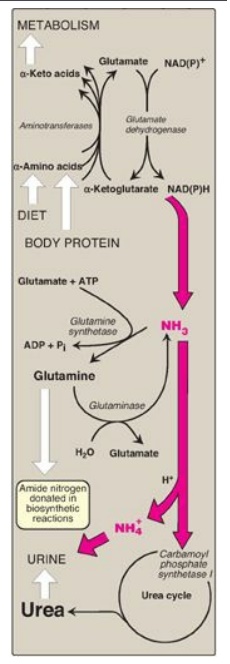
Figure 19.19 Metabolism of ammonia
(NH3). [Note: Glutamate dehydrogenase is one of several sources of NH3.]
Urea content in the urine is reported as urinary urea nitrogen, or UUN. Urea in
blood is reported as BUN (blood urea nitrogen). The enzymes glutamate
dehydrogenase, glutamine synthetase, and carbamoyl phosphate synthetase I fix NH3
into organic molecules.
C. Hyperammonemia
The capacity of the
hepatic urea cycle exceeds the normal rates of ammonia generation, and the
levels of serum ammonia are normally low (5–35 µmol/l). However, when liver
function is compromised, due either to genetic defects of the urea cycle or
liver disease, blood levels can rise above 1,000 µmol/l. Such hyperammonemia is
a medical emergency, because ammonia has a direct neurotoxic effect on the CNS.
For example, elevated concentrations of ammonia in the blood cause the symptoms
of ammonia intoxication, which include tremors, slurring of speech, somnolence
(drowsiness), vomiting, cerebral edema, and blurring of vision. At high
concentrations, ammonia can cause coma and death. There are two major types of
hyperammonemia.
1. Acquired hyperammonemia: Liver disease is a common cause of
hyperammonemia in adults and may be due, for example, to viral hepatitis or to
hepatotoxins such as alcohol. Cirrhosis of the liver may result in formation of
collateral circulation around the liver. As a result, portal blood is shunted
directly into the systemic circulation and does not have access to the liver.
Therefore, the conversion of ammonia to urea is severely impaired, leading to
elevated levels of ammonia.
2. Congenital hyperammonemia: Genetic deficiencies of each of
the five enzymes of the urea cycle have been described, with an overall
incidence estimated to be 1:25,000 live births. X-linked ornithine
transcarbamoylase deficiency is the most common of these disorders,
predominantly affecting males, although female carriers may become symptomatic.
All of the other urea cycle disorders follow an autosomal-recessive inheritance
pattern. In each case, the failure to synthesize urea leads to hyperammonemia
during the first weeks following birth. [Note: The hyperammonemia seen with
arginase deficiency is less severe because arginine contains two waste
nitrogens and can be excreted in the urine.] Historically, urea cycle defects had
high morbidity (neurologic manifestations) and mortality. Treatment included
restriction of dietary protein in the presence of sufficient calories to
prevent catabolism. Administration of compounds that bind covalently to amino
acids, producing nitrogen-containing molecules that are excreted in the urine,
has improved survival. For example, phenylbutyrate given orally is converted to
phenylacetate. This condenses with glutamine to form phenylacetylglutamine,
which is excreted (Figure 19.20).
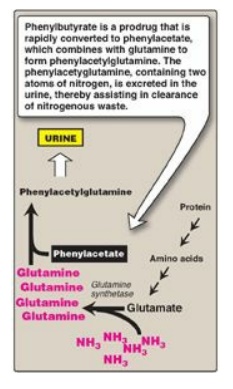
Figure 19.20 Treatment of
patients with urea cycle defects by administration of phenylbutyrate to aid in
excretion of ammonia (NH3).
Related Topics
