Catabolism of the Carbon Skeletons of Amino Acids
| Home | | Biochemistry |Chapter: Biochemistry : Amino Acid Degradation and Synthesis
The pathways by which amino acids are catabolized are conveniently organized according to which one (or more) of the seven intermediates listed above is produced from a particular amino acid.
CATABOLISM OF THE CARBON SKELETONS OF AMINO ACIDS
The pathways by which
amino acids are catabolized are conveniently organized according to which one
(or more) of the seven intermediates listed above is produced from a particular
amino acid.
A. Amino acids that form oxaloacetate
Asparagine is hydrolyzed by asparaginase, liberating ammonium (NH4+) and aspartate (Figure 20.3). Aspartate loses its amino group by transamination to form oxaloacetate (see Figure 20.3). [Note: Some rapidly dividing leukemic cells are unable to synthesize sufficient asparagine to support their growth. This makes asparagine an essential amino acid for these cells, which, therefore, require asparagine from the blood. Asparaginase, which hydrolyzes asparagine to aspartate, can be administered systemically to treat leukemic patients. Asparaginase lowers the level of asparagine in the plasma, thereby depriving cancer cells of a required nutrient.]
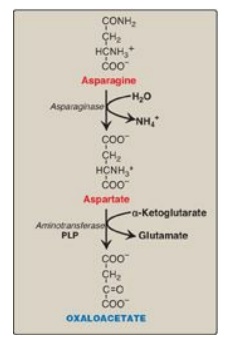
Figure 20.3 Metabolism of asparagine and aspartate. [Note: Recall that carbons from aspartate give rise to fumarate in the urea cycle.] PLP = pyridoxal phosphate.
B. Amino acids that form α-ketoglutarate via glutamate
1. Glutamine: This amino acid is hydrolyzed to glutamate and
ammonium by the enzyme glutaminase. Glutamate is converted to α-ketoglutarate
by transamination or through oxidative deamination by glutamate dehydrogenase.
2. Proline: This amino acid is oxidized to glutamate. Glutamate
is transaminated or oxidatively deaminated to form α-ketoglutarate.
3. Arginine: This amino acid is hydrolyzed by arginase to
produce ornithine (and urea). [Note: This reaction occurs primarily in the
liver as part of the urea cycle .] Ornithine is subsequently converted to α-ketoglutarate,
with glutamate semialdehyde as an intermediate.
4. Histidine: This amino acid is oxidatively deaminated by histidase to urocanic acid, which subsequently forms N-formiminoglutamate ([FIGlu] Figure 20.4). FIGlu donates its formimino group to tetrahydrofolate (THF), leaving glutamate, which is degraded as described above. [Note: Individuals deficient in folic acid excrete increased amounts of FIGlu in the urine, particularly after ingestion of a large dose of histidine. The FIGlu excretion test has been used in diagnosing a deficiency of folic acid.]
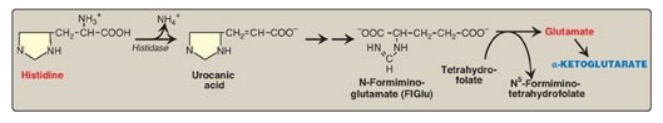
Figure 20.4 Degradation of histidine.
C. Amino acids that form pyruvate
1. Alanine: This amino acid loses its amino group by transamination to form pyruvate (Figure 20.5). [Note: Alanine is the major gluconeogenic amino acid.]
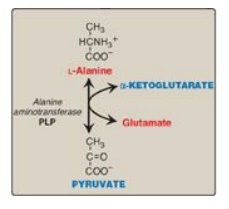
Figure 20.5 Transamination of alanine to form pyruvate. PLP = pyridoxal phosphate.
2. Serine: This amino acid can be converted to glycine and
N5,N10-methylenetetrahydrofolate (Figure 20.6A). Serine can also be converted
to pyruvate by serine dehydratase (Figure 20.6B).
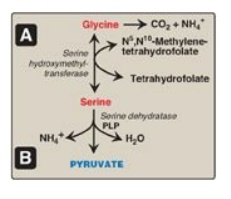
Figure 20.6 A. Interconversion of serine and glycine and oxidation of glycine. B. Dehydration of serine to form pyruvate. PLP = pyridoxal phosphate.
3. Glycine: This amino acid can be converted to serine by the
reversible addition of a methylene group from N5,N10-methylenetetrahydrofolic
acid (see Figure 20.6A) or oxidized to CO2 and NH4+.
[Note: Glycine can be deaminated to glyoxylate, which can be oxidized to
oxalate or transaminated to glycine. Deficiency of the transaminase in liver
peroxisomes causes overproduction of oxalate, the formation of oxalate stones,
and kidney damage (primary oxaluria type 1).]
4. Cysteine: This amino acid undergoes desulfuration to yield
pyruvate. [Note: The sulfate released can be used to synthesize
3-phosphoadenosine-5I-phosphosulfate
(PAPS), an activated sulfur donor to a variety of acceptors.] Cysteine can be
oxidized to its disulfide derivative, cystine.
5. Threonine: This amino acid is converted to pyruvate in most organisms but is a minor pathway (at best) in humans.
D. Amino acids that form fumarate
1. Phenylalanine and tyrosine: Hydroxylation of phenylalanine
produces tyrosine (Figure 20.7). This reaction, catalyzed by
tetrahydrobiopterin-requiring phenylalanine hydroxylase, initiates the
catabolism of phenylalanine. Thus, the metabolism of phenylalanine and tyrosine
merge, leading ultimately to the formation of fumarate and acetoacetate.
Phenylalanine and tyrosine are, therefore, both glucogenic and ketogenic.
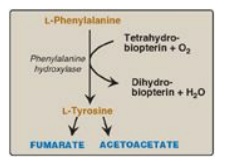
Figure 20.7 Degradation of phenylalanine.
2. Inherited deficiencies: Inherited deficiencies in the enzymes of phenylalanine and tyrosine metabolism lead to the diseases phenylketonuria, tyrosimenia, and alkaptonuria as well as the condition of albinism.
E. Amino acids that form succinyl coenzyme A: methionine
Methionine is one of
four amino acids that form succinyl CoA. This sulfur-containing amino acid
deserves special attention because it is converted to S-adenosylmethionine
(SAM), the major methyl-group donor in one-carbon metabolism (Figure 20.8).
Methionine is also the source of homocysteine, a metabolite associated with
atherosclerotic vascular disease and thrombosis.
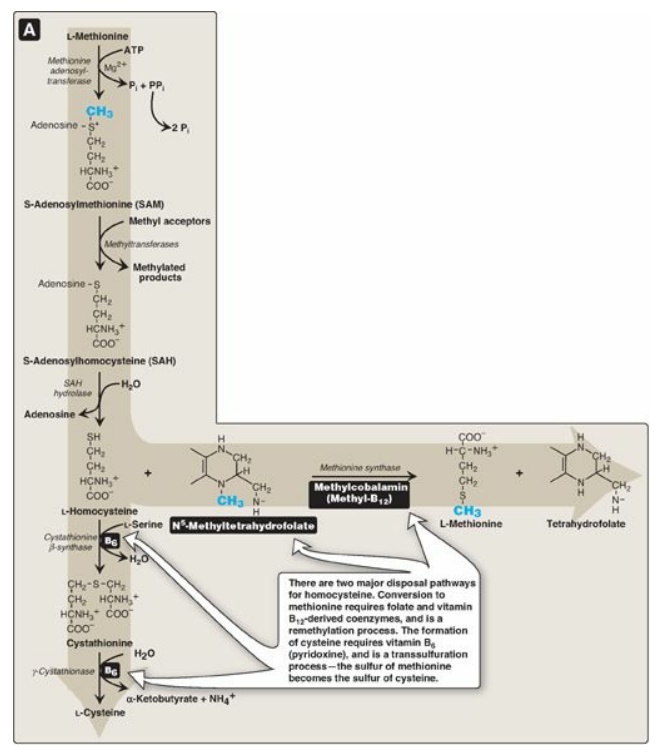
Figure 20.8 Degradation and resynthesis of methionine. [Note: The resynthesis of methionine from homocysteine is the only reaction in which tetrahydrofolate both carries and donates a methyl (-CH3) group. In all other reactions, SAM is the methyl group carrier and donor.] PPi = pyrophosphate; Pi = inorganic phosphate.
1. Synthesis of S-adenosylmethionine: Methionine condenses with adenosine triphosphate (ATP), forming SAM, a high-energy compound that is unusual in that it contains no phosphate. The formation of SAM is driven, in effect, by hydrolysis of all three phosphate bonds in ATP (see Figure 20.8).
2. Activated methyl group: The methyl group attached to the
tertiary sulfur in SAM is “activated” and can be transferred by
methyltransferases to a variety of acceptor molecules such as norepinephrine in
the synthesis of epinephrine. The methyl group is usually transferred to
nitrogen (as with epinephrine) or oxygen atoms (as with the catechols;) and
sometimes to carbon atoms (as with cytosine). The reaction product,
S-adenosylhomocysteine (SAH), is a simple thioether, analogous to methionine.
The resulting loss of free energy makes methyl transfer essentially
irreversible.
3. Hydrolysis of S-adenosylhomocysteine: After donation of the methyl group,
SAH is hydrolyzed to homocysteine (Hcy) and adenosine. Hcy has two fates. If
there is a deficiency of methionine, Hcy may be remethylated to methionine (see
Figure 20.8). If methionine stores are adequate, Hcy may enter the
transsulfuration pathway, where it is converted to cysteine.
a. Resynthesis of methionine: Hcy accepts a methyl group from
N5-methyltetrahydrofolate (N5-methyl-THF) in a reaction requiring
methylcobalamin, a coenzyme derived from vitamin B12. [Note: The
methyl group is transferred by methionine synthase from the B12
derivative to Hcy, regenerating methionine. Cobalamin is remethylated from N5-methyl-THF.]
b. Synthesis of cysteine: Hcy condenses with serine, forming cystathionine, which is hydrolyzed to α-ketobutyrate and cysteine (see Figure 20.8). This vitamin B6–requiring sequence has the net effect of converting serine to cysteine and Hcy to α-ketobutyrate, which is oxidatively decarboxylated to form propionyl CoA. Propionyl CoA is converted to succinyl CoA. Because Hcy is synthesized from the essential amino acid methionine, cysteine is not an essential amino acid as long as sufficient methionine is available.
4. Relationship of homocysteine to vascular
disease: Elevations
in plasma Hcy levels promote oxidative damage, inflammation, and endothelial
dysfunction and are an independent risk factor for occlusive vascular disease
(Figure 20.9). Mild elevations are seen in about 7% of the population.
Epidemiologic studies have shown that plasma Hcy levels are inversely related
to plasma levels of folate, B12, and B6, the three vitamins involved
in the conversion of Hcy to methionine or cysteine. Supplementation with these
vitamins has been shown to reduce circulating levels of Hcy. However, in
patients with established cardiovascular disease, vitamin therapy does not
decrease cardiovascular events or death. This raises the question as to whether
Hcy is a cause of the vascular damage or merely a marker of such damage. [Note:
Large elevations in plasma Hcy as a result of rare deficiencies in
cystathionine β-synthase of the transsulfuration pathway are seen in patients
with classic homocystinuria. These individuals experience premature vascular
disease, with about 25% dying from thrombotic complications before age 30
years.] Deficiencies in the remethylation reaction also result in a rise in
Hcy.
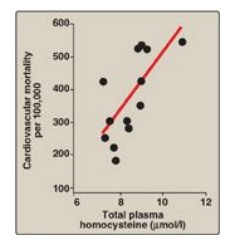
Figure 20.9 Association between cardiovascular disease mortality and total plasma homocysteine.
Elevated homocysteine and decreased folic acid
levels in pregnant women are associated with increased incidence of neural tube
defects (improper closure, as in spina bifida) in the fetus. Periconceptual
supplementation with folate reduces the risk of such defects.
F. Other amino acids that form succinyl coenzyme A
Degradation of valine,
isoleucine, and threonine also results in the production of succinyl CoA, a TCA
cycle intermediate and glucogenic compound.
1. Valine and isoleucine: These amino acids are branched-chain amino acids (BCAAs) that generate propionyl CoA, which is converted to methylmalonyl CoA and then succinyl CoA by biotin- and vitamin B12–requiring reactions (Figure 20.10).
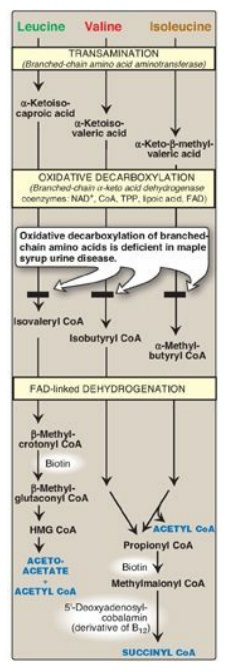
Figure 20.10 Degradation of leucine, valine, and isoleucine. [Note: β-Methylcrotonyl CoA carboxylase is one of four biotinrequiring carboxylases we have encountered. The other three are pyruvate carboxylase, acetyl CoA carboxylase, and propionyl CoA carboxylase.] TPP = thiamine pyrophosphate; FAD = flavin adenine dinucleotide; CoA = coenzyme A; NAD = nicotinamide adenine dinucleotide; HMG = hydroxymethylglutarate.
2. Threonine: This amino acid is dehydrated to α-ketobutyrate,
which is converted to propionyl CoA and then to succinyl CoA. Propionyl CoA,
then, is generated by the catabolism of the amino acids methionine, valine,
isoleucine, and threonine. [Note: Propionyl CoA also is generated by the
oxidation of odd-numbered fatty acids.]
G. Amino acids that form acetyl coenzyme A or acetoacetyl coenzyme A
Leucine, isoleucine,
lysine, and tryptophan form acetyl CoA or acetoacetyl CoA directly, without
pyruvate serving as an intermediate (through the pyruvate dehydrogenase
reaction;). As noted earlier, phenylalanine and tyrosine also give rise to
acetoacetate during their catabolism (see Figure 20.7). Therefore, there are a
total of six partly or wholly ketogenic amino acids.
1. Leucine: This amino acid is exclusively ketogenic in its
catabolism, forming acetyl CoA and acetoacetate (see Figure 20.10). The initial
steps in the catabolism of leucine are similar to those of the other BCAAs,
isoleucine and valine (see below).
2. Isoleucine: This amino acid is both ketogenic and glucogenic,
because its metabolism yields acetyl CoA and propionyl CoA. The first three
steps in the metabolism of isoleucine are similar to the initial steps in the
degradation of the other BCAAs, valine and leucine (see Figure 20.10).
3. Lysine: This amino acid is exclusively ketogenic and is
unusual in that neither of its amino groups undergoes transamination as the
first step in catabolism. Lysine is ultimately converted to acetoacetyl CoA.
4. Tryptophan: This amino acid is both glucogenic and ketogenic because its catabolism yields alanine and acetoacetyl CoA. [Note: Quinolinate from tryptophan catabolism is used in the synthesis of nicotinamide adenine dinucleotide.]
H. Catabolism of the branched-chain amino acids
The BCAAs isoleucine,
leucine, and valine are essential amino acids. In contrast to other amino
acids, they are metabolized primarily by the peripheral tissues (particularly
muscle), rather than by the liver. Because these three amino acids have a
similar route of catabolism, it is convenient to describe them as a group (see
Figure 20.10).
1. Transamination: Transfer of the amino groups of all
three BCAAs to α - ketoglutarate is catalyzed by a single, vitamin B6–requiring
enzyme, branched-chain amino acid aminotransferase.
2. Oxidative decarboxylation: Removal of the carboxyl group of
the α-keto acids derived from leucine, valine, and isoleucine is catalyzed by a
single multienzyme complex, branched-chain α-keto acid dehydrogenase (BCKD)
complex. This complex uses thiamine pyrophosphate, lipoic acid, flavin adenine
dinucleotide (FAD), NAD+, and CoA as its coenzymes and produces
NADH. [Note: This reaction is similar to the conversion of pyruvate to acetyl
CoA by pyruvate dehydrogenase (PDH) complex and the oxidation of
α-ketoglutarate to succinyl CoA by α-ketoglutarate dehydrogenase complex. The
Enzyme 3 (E3) component is identical in BCKD, PDH, and α-ketoglutarate
dehydrogenase.]
3. Dehydrogenation: Oxidation of the products formed
in the BCKD reaction yields α-β-unsaturated acyl CoA derivatives and FADH2.
This reaction is analogous to the FAD-linked dehydrogenation in the β-oxidation
of fatty acids. [Note: Deficiency in the dehydrogenase specific for isovaleryl
CoA causes neurologic problems and is associated with a “sweaty feet” odor in
body fluids.]
4. End products: The catabolism of isoleucine ultimately yields
acetyl CoA and succinyl CoA, rendering it both ketogenic and glucogenic. Valine
yields succinyl CoA and is glucogenic. Leucine is ketogenic, being metabolized
to acetoacetate and acetyl CoA. In addition, NADH and FADH2 are produced
in the decarboxylation and dehydrogenation reactions, respectively. [Note: BCAA
catabolism also results in glutamine and alanine being sent out into the blood
from muscle.]
Related Topics
