Porphyrin Metabolism
| Home | | Biochemistry |Chapter: Biochemistry : Conversion of Amino Acids to Specialized Products
Porphyrins are cyclic compounds that readily bind metal ions, usually ferrous (Fe 2+) or ferric (Fe3+) iron.
PORPHYRIN METABOLISM
Porphyrins are cyclic
compounds that readily bind metal ions, usually ferrous (Fe 2+) or ferric
(Fe3+) iron. The most prevalent metalloporphyrin in humans is heme, which
consists of one Fe2+ coordinated in the center of the tetrapyrrole ring of
protoporphyrin IX. Heme is the prosthetic group for hemoglobin, myoglobin, the
cytochromes, the cytochrome P450 (CYP) monooxygenase system, catalase, nitric
oxide synthase, and peroxidase. These hemeproteins are rapidly synthesized and
degraded. For example, 6–7 g of hemoglobin are synthesized each day to replace
heme lost through the normal turnover of erythrocytes. The simultaneous
synthesis and degradation of the associated porphyrins and recycling of the
bound iron ions is coordinated with the turnover of hemeproteins.
A. Structure of porphyrins
Porphyrins are cyclic
molecules formed by the linkage of four pyrrole rings through methenyl bridges
(Figure 21.2). Three structural features of these molecules are relevant to
understanding their medical significance.
1. Side chains: Different porphyrins vary in the
nature of the side chains that are attached to each of the four pyrrole rings.
Uroporphyrin contains acetate (–CH2– COO–) and propionate (–CH2–CH2–COO–)
side chains; coproporphyrin contains methyl (–CH3) and propionate groups; and
protoporphyrin IX (and heme) contains vinyl (–CH=CH2), methyl, and
propionate groups.
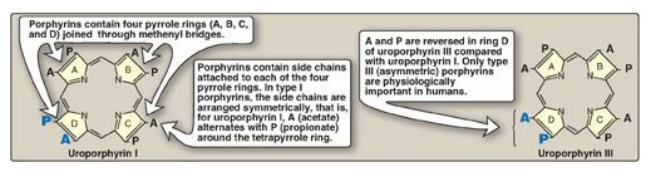
Figure 21.2 Structures of
uroporphyrin I and uroporphyrin III.
2. Distribution of side chains: The side chains
of porphyrins can be ordered around the tetrapyrrole nucleus in four different
ways, designated by Roman numerals I to IV. Only type III porphyrins, which
contain an asymmetric substitution on ring D (see Figure 21.2), are
physiologically important in humans. [Note: Protoporphyrin IX is a member of
the type III series.]
3. Porphyrinogens: These porphyrin precursors
(for example, uroporphyrinogen) exist in a chemically reduced, colorless form
and serve as intermediates between porphobilinogen and the oxidized, colored
protoporphyrins in heme biosynthesis.
B. Biosynthesis of heme
The major sites of heme
biosynthesis are the liver, which synthesizes a number of heme proteins
(particularly the CYP proteins), and the erythrocyte-producing cells of the
bone marrow, which are active in hemoglobin synthesis. [Note: Over 85% of all
heme synthesis occurs in erythroid tissue.] In the liver, the rate of heme
synthesis is highly variable, responding to alterations in the cellular heme
pool caused by fluctuating demands for heme proteins. In contrast, heme
synthesis in erythroid cells is relatively constant and is matched to the rate
of globin synthesis. The initial reaction and the last three steps in the
formation of porphyrins occur in mitochondria, whereas the intermediate steps
of the biosynthetic pathway occur in the cytosol (see Figure 21.8). [Note:
Mature red blood cells (RBCs) lack mitochondria and are unable to synthesize
heme.]
1. Formation of δ-aminolevulinic acid: All the
carbon and nitrogen atoms of the porphyrin molecule are provided by glycine (a
nonessential amino acid) and succinyl coenzyme A (a citric acid cycle
intermediate) that condense to form δ-aminolevulinic acid (ALA) in a reaction
catalyzed by ALA synthase ([ALAS] Figure 21.3) This reaction requires pyridoxal
phosphate (PLP) as a coenzyme and is the committed and rate-limiting step in
porphyrin biosynthesis. [Note: There are two isoforms of ALAS, 1 and 2, each
produced by different genes and controlled by different mechanisms. ALAS1 is
found in all tissues, whereas ALAS2 is erythroid specific. Loss-of-function
mutations in ALAS2 result in X-linked sideroblastic anemia.]
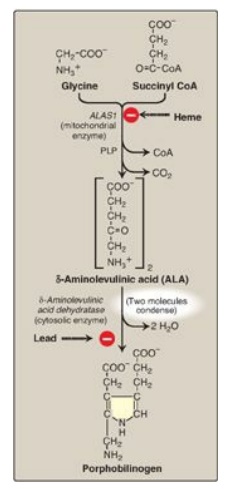
Figure 21.3 Pathway of porphyrin synthesis: Formation of porphobilinogen. ALAS = δ-aminolevulinic acid synthase; CoA = coenzyme A; PLP = pyridoxal phosphate. (Continued in Figures 21.4 and 21.5.)
a. Effect of heme (hemin): When porphyrin production exceeds the availability of the apoproteins that require it, heme accumulates and is converted to hemin by the oxidation of Fe2+ to Fe3+. Hemin decreases the amount (and thereby the activity) of ALAS1 by repressing transcription of its gene, increasing degradation of its messenger RNA, and decreasing import of the enzyme into mitochondria. [Note: In erythroid cells, ALAS2 is controlled by the availability of intracellular iron.]
b. Effect of drugs: Administration of any of a large number of drugs results in a significant increase in hepatic ALAS1 activity. These drugs are metabolized by t h e microsomal CYP monooxygenase system, a hemeprotein oxidase system found in the liver. In response to these drugs, the synthesis of CYP proteins increases, leading to an enhanced consumption of heme, a component of these proteins. This, in turn, causes a decrease in the concentration of heme in liver cells. The lower intracellular heme concentration leads to an increase in the synthesis of ALAS1 and prompts a corresponding increase in the synthesis of ALA.
2. Formation of porphobilinogen: The condensation
of two molecules of ALA to form porphobilinogen by zinc-containing ALA
dehydratase (porphobilinogen synthase) is extremely sensitive to inhibition by
heavy metal ions (for example, lead) that replace the zinc (see Figure 21.3).
This inhibition is, in part, responsible for the elevation in ALA and the
anemia seen in lead poisoning.
3. Formation of uroporphyrinogen: The condensation of four porphobilinogens produces the linear tetrapyrrole hydroxymethylbilane, which is cyclized and isomerized by uroporphyrinogen III synthase to produce the asymmetric uroporphyrinogen III. This cyclic tetrapyrrole undergoes decarboxylation of its acetate groups, generating coproporphyrinogen III (Figure 21.4). These reactions occur in the cytosol.
4. Formation of heme: Coproporphyrinogen III
enters the mitochondrion, and two propionate side chains are decarboxylated by
coproporphyrinogen III oxidase to vinyl groups generating protoporphyrinogen
IX, which is oxidized to protoporphyrin IX. The introduction of iron (as Fe2+)
into protoporphyrin IX can occur spontaneously, but the rate is enhanced by
ferrochelatase, an enzyme that, like ALA dehydratase, is inhibited by lead
(Figure 21.5).
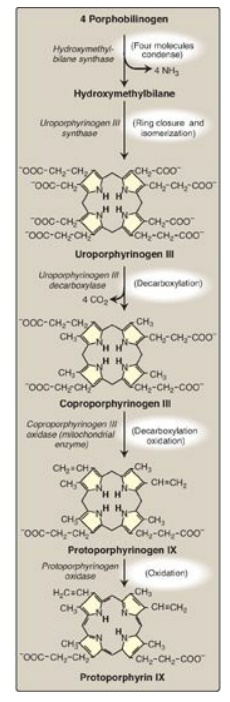
Figure 21.4 Pathway of porphyrin synthesis: formation of protoporphyrin IX. (Continued from Figure 21.3.) [Note: Deficiency in uroporphyrinogen III synthase prevents isomerization, resulting in production of type I porphyrins.]
C. Porphyrias
Porphyrias are rare,
inherited (or occasionally acquired) defects in heme synthesis, resulting in
the accumulation and increased excretion of porphyrins or porphyrin precursors
(see Figure 21.8). [Note: With few exceptions, porphyrias are inherited as
autosomal-dominant disorders.] Each porphyria results in the accumulation of a
unique pattern of intermediates caused by the deficiency of an enzyme in the
heme synthetic pathway. [Note: “Porphyria” refers to the red-blue color caused
by pigment-like porphyrins in the urine of some patients with defects in heme
synthesis.]
Clinical manifestations: The porphyrias are classified as erythropoietic or hepatic, depending on whether the enzyme deficiency occurs in the erythropoietic cells of the bone marrow or in the liver. Hepatic porphyrias can be further classified as chronic or acute. In general, individuals with an enzyme defect prior to the synthesis of the tetrapyrroles manifest abdominal and neuropsychiatric signs, whereas those with enzyme defects leading to the accumulation of tetrapyrrole intermediates show photosensitivity (that is, their skin itches and burns [pruritus] when exposed to visible light). [Note: Photosenstivity is a result of the oxidation of colorless porphyrinogens to colored porphyrins, which are photosensitizing molecules thought to participate in the formation of superoxide radicals from oxygen. These reactive oxygen species can oxidatively damage membranes and cause the release of destructive enzymes from lysosomes.]
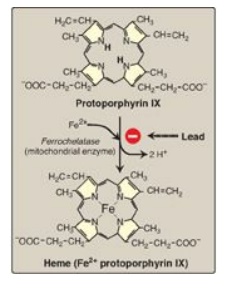
Figure 21.5 Pathway of
porphyrin synthesis: formation of heme. (Continued from Figures 21.3 and 21.4.)
Fe2+ = ferrous iron.
a. Chronic hepatic
porphyria: Porphyria cutanea tarda, the most common porphyria, is a chronic
disease of the liver. The disease is associated with a deficiency in
uroporphyrinogen decarboxylase, but clinical expression of the enzyme
deficiency is influenced by various factors, such as hepatic iron overload,
exposure to sunlight, alcohol ingestion, estrogen therapy, and the presence of
hepatitis B or C or HIV infections. Clinical onset is typically during the
fourth or fifth decade of life. Porphyrin accumulation leads to cutaneous
symptoms (Figure 21.6) as well as urine that is red to brown in natural light
(Figure 21.7) and pink to red in fluorescent light.
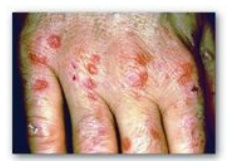
Figure 21.6 Skin eruptions in
a patient with porphyria cutanea tarda.

Figure 21.7 Urine from a patient with porphyria cutanea tarda (right) and from a patient with normal porphyrin excretion (left).
b. Acute hepatic porphyrias: Acute hepatic porphyrias (ALA dehydratase deficiency porphyria, acute intermittent porphyria, hereditary coproporphyria, and variegate porphyria) are characterized by acute attacks of gastrointestinal (GI), neuropsychiatric, and motor symptoms that may be accompanied by photosensitivity. Porphyrias leading to accumulation of ALA and porphobilinogen, such as acute intermittent porphyria, cause abdominal pain and neuropsychiatric disturbances, ranging from anxiety to delirium. Symptoms of the acute hepatic porphyrias are often precipitated by use of drugs, such as barbiturates and ethanol, which induce the synthesis of the heme-containing CYP microsomal drug oxidation system. This further decreases the amount of available heme, which, in turn, promotes increased synthesis of ALAS1.
c. Erythropoietic
porphyrias: The chronic erythropoietic porphyrias (congenital erythropoietic
porphyria and erythropoietic protoporphyria) are characterized by skin rashes
and blisters that appear in early childhood. [Note: Patients with
erythropoietic protoporphyria are also at risk for hepatobiliary disease.]
2. Increased δ-aminolevulinic acid synthase
activity: One common feature of the porphyrias is a decreased synthesis of
heme. In the liver, heme normally functions as a repressor of the gene for
ALAS1. Therefore, the absence of this end product results in an increase in the
synthesis of ALAS1 (derepression). This causes an increased synthesis of
intermediates that occur prior to the genetic block. The accumulation of these
toxic intermediates is the major pathophysiology of the porphyrias.
3. Treatment: During acute porphyria attacks,
patients require medical support, particularly treatment for pain and vomiting.
The severity of acute symptoms of the porphyrias can be diminished by
intravenous injection of hemin and glucose, which decreases the synthesis of
ALAS1. Protection from sunlight, ingestion of β-carotene (a free-radical
scavenger), and phlebotomy are helpful in porphyrias with photosensitivity.
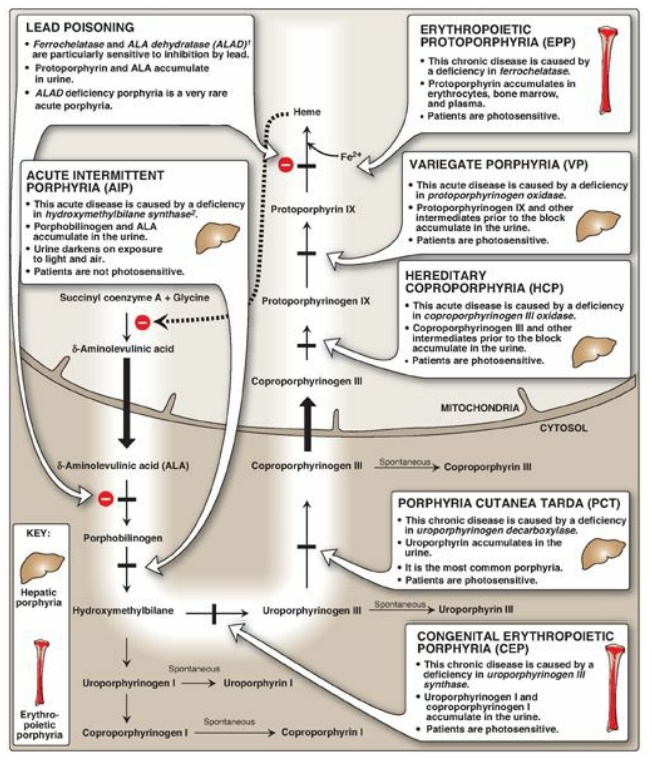
Figure 21.8 Summary of heme
synthesis. 1Also referred to as porphobilinogen synthase. 2Also referred to as
porphobilinogen deaminase. [Note: Deficiencies in ALA synthase-1 (ALAS1) are
unknown. Deficiencies in ALAS2 result in an anemia, not a porphyria.]
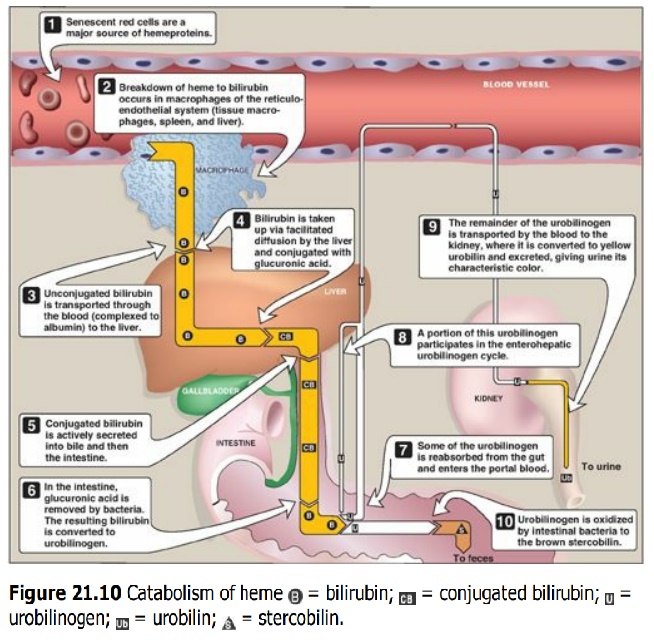
D. Degradation of heme
After approximately 120
days in the circulation, red blood cells are taken up and degraded by the
reticuloendothelial system, particularly in the liver and spleen (Figure 21.9).
Approximately 85% of heme destined for degradation comes from senescent RBCs.
The remainder is from the degradation of heme proteins other than hemoglobin.
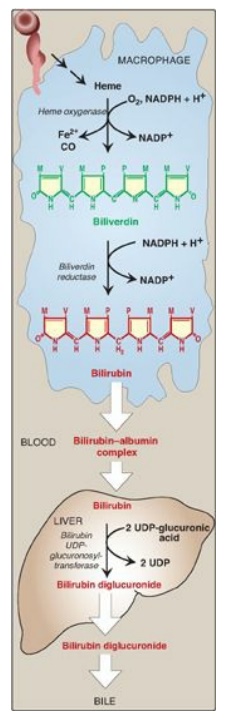
Figure 21.9 Formation of bilirubin from heme and its conversion to bilirubin diglucuronide. UDP = uridine diphosphate; CO = carbon monoxide; NADP(H) = nicotinamide adenine dinucleotide phosphate.
1. Formation of bilirubin: The first step in the degradation of heme is catalyzed by the microsomal heme oxygenase system of the reticuloendothelial cells. In the presence of nicotinamide adenine dinucleotide phosphate and O2, the enzyme catalyzes three successive oxygenations that result in opening of the porphyrin ring (converting cyclic heme to linear biliverdin), production of carbon monoxide (CO), and release of Fe2+ (see Figure 21.9).
[Note: The CO has biologic
function, acting as a signaling molecule and anti-inflammatory.] Biliverdin, a
green pigment, is reduced, forming the red-orange bilirubin. Bilirubin and its
derivatives are collectively termed bile pigments. [Note: The changing colors
of a bruise reflect the varying pattern of intermediates that occurs during
heme degradation.]
Bilirubin, unique to
mammals, appears to function at low levels as an antioxidant. In this role, it
is oxidized to biliverdin, which is then reduced by biliverdin reductase,
regenerating bilirubin.
2. Uptake of bilirubin by the liver: Bilirubin is
only slightly soluble in plasma and, therefore, is transported to the liver by
binding noncovalently to albumin. [Note: Certain anionic drugs, such as
salicylates and sulfonamides, can displace bilirubin from albumin, permitting
bilirubin to enter the central nervous system (CNS). This causes the potential
for neural damage in infants.] Bilirubin dissociates from the carrier albumin
molecule; enters a hepatocyte via facilitated diffusion; and binds to
intracellular proteins, particularly the protein ligandin.
3. Formation of bilirubin diglucuronide: In the
hepatocyte, the solubility of bilirubin is increased by the addition of two
molecules of glucuronic acid, producing bilirubin diglucuronide. [Note: This
process is referred to as conjugation.] The reaction is catalyzed by microsomal
bilirubin UDP-glucuronosyltransferase (bilirubin UGT) using uridine diphosphate
(UDP)-glucuronic acid as the glucuronate donor. [Note: Varying degrees of
deficiency of bilirubin UGT result in Crigler-Najjar I and II and Gilbert
syndrome, with Crigler-Najjar I being the most severe deficiency.]
4. Secretion of bilirubin into bile: Bilirubin
diglucuronide (conjugated bilirubin [CB]) is actively transported against a
concentration gradient into the bile canaliculi and then into the bile. This
energy-dependent, rate-limiting step is susceptible to impairment in liver
disease. [Note: A rare deficiency in the protein required for transport of CB
out of the liver results in Dubin-Johnson syndrome.] Unconjugated bilirubin
(UCB) is normally not secreted into bile.
5. Formation of urobilins in the intestine:
Bilirubin diglucuronide is hydrolyzed and reduced by bacteria in the gut to
yield urobilinogen, a colorless compound. Most of the urobilinogen is oxidized
by intestinal bacteria to stercobilin, which gives feces the characteristic
brown color. However, some of the urobilinogen is reabsorbed from the gut and
enters the portal blood. A portion of this urobilinogen participates in the
enterohepatic urobilinogen cycle in which it is taken up by the liver and then
resecreted into the bile. The remainder of the urobilinogen is transported by
the blood to the kidney, where it is converted to yellow urobilin and excreted,
giving urine its characteristic color. The metabolism of bilirubin is
summarized in Figure 21.10.
E. Jaundice
Jaundice (also called
icterus) refers to the yellow color of skin, nail beds, and sclerae (whites of
the eyes) caused by deposition of bilirubin, secondary to increased bilirubin
levels in the blood (hyper-bilirubinemia) as shown in Figure 21.11. Although
not a disease, jaundice is usually a symptom of an underlying disorder. [Note:
Blood bilirubin levels are normally about 1 mg/dl. Jaundice is seen at 2–3
mg/dl.]
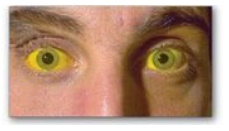
Figure 21.11 Jaundiced
patient, with the sclerae of his eyes appearing yellow.
1. Types of jaundice: Jaundice can be classified
into three major types described below. However, in clinical practice, jaundice
is often more complex than indicated in this simple classification. For
example, the accumulation of bilirubin may be a result of defects at more than
one step in its metabolism.
a. Hemolytic jaundice: The liver has the capacity to conjugate and excrete over 3,000 mg of bilirubin per day, whereas the normal production of bilirubin is only 300 mg/day. This excess capacity allows the liver to respond to increased heme degradation with a corresponding increase in conjugation and secretion of bilirubin diglucuronide.
However, extensive hemolysis (for example, in patients with sickle cell anemia
or pyruvate kinase or glucose 6-phosphate dehydrogenase deficiency) may produce
bilirubin faster than it can be conjugated. UCB levels in the blood become
elevated, causing jaundice (Figure 21.12A). [Note: With hemolysis, more CB is made
and excreted into the bile, the amount of urobilinogen entering the
enterohepatic circulation is increased, and urinary urobilinogen is increased.]
b. Hepatocellular
jaundice: Damage to liver cells (for example, in patients with cirrhosis or
hepatitis) can cause UCB levels in the blood to increase as a result of
decreased conjugation. Urobilinogen is increased in the urine because hepatic
damage decreases the enterohepatic circulation of this compound, allowing more
to enter the blood, from which it is filtered into the urine. The urine
consequently darkens, whereas stools may be a pale, clay color. Plasma levels
of alanine and aspartate transaminases (AST and ALT, respectively;) are
elevated. [Note: If CB is made but is not efficiently secreted from the liver
into bile (intrahepatic cholestasis), it can diffuse (“leak”) into the blood,
causing a conjugated hyperbilirubinemia.]
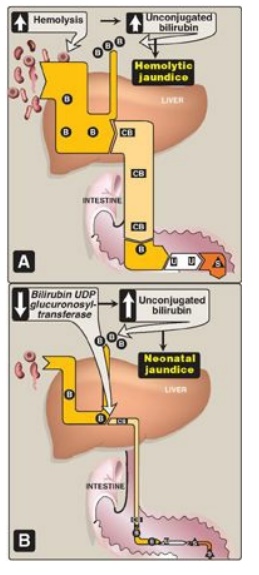
Figure 21.12 Alterations in
the metabolism of heme. A. Hemolytic jaundice. B. Neonatal jaundice. CB= conjugated
bilirubin; B= bilirubin; u= urobilinogen; s= stercobilin; UDP = uridine
diphosphate
C. Obstructive
jaundice: In this instance, jaundice is not caused by overproduction of
bilirubin or decreased conjugation but, instead, results from obstruction of
the common bile duct (extrahepatic cholestasis). For example, the presence of a
tumor or bile stones may block the duct, preventing passage of CB into the
intestine. Patients with obstructive jaundice experience GI pain and nausea and
produce stools that are a pale, clay color. The liver “regurgitates” CB into
the blood (hyperbilirubinemia). The CB is eventually excreted in the urine
(which darkens upon standing), and is referred to as “urinary bilirubin.”
Urinary urobilinogen is absent.
2. Jaundice in newborns: The majority of newborn
infants (60% of full term and 80% of preterm) show a rise in UCB in the first
postnatal week (and a transient, “physiologic” jaundice) because the activity
of hepatic bilirubin UGT is low at birth (it reaches adult levels in about 4
weeks) as shown in Figures 21.12B and 21.13. Elevated UCB, in excess of the
binding capacity of albumin (20–25 mg/dl), can diffuse into the basal ganglia,
cause toxic encephalopathy (kernicterus) and a pathologic jaundice. Therefore,
newborns with significantly elevated bilirubin levels are treated with blue
fluorescent light (phototherapy), as shown in Figure 21.14, which converts
bilirubin to more polar and, therefore, water-soluble isomers. These
photoisomers can be excreted into the bile without conjugation to glucuronic
acid. [Note: Because of solubility differences, only UCB crosses the blood
brain barrier, and only CB appears in urine.]
3. Determination of bilirubin concentration: Bilirubin is commonly measured by the van den Bergh reaction, in which diazotized sulfanilic acid reacts with bilirubin to form red azodipyrroles that are measured colorimetrically.
In aqueous solution, the
water-soluble CB reacts rapidly with the reagent (within one minute), and is
said to be “direct-reacting.” The UCB, which is much less soluble in aqueous
solution, reacts more slowly. However, when the reaction is carried out in
methanol, both CB and UCB are soluble and react with the reagent, providing the
total bilirubin value. The “indirect-reacting” bilirubin, which corresponds to
the UCB, is obtained by subtracting the direct-reacting bilirubin from the
total bilirubin. [Note: In normal plasma, only about 4% of the total bilirubin
is conjugated or direct-reacting, because most is secreted into bile.]
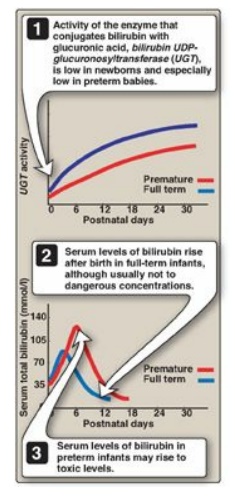
Figure 21.13 Neonatal
jaundice. UDP = uridine diphosphate.
Figure 21.14 Phototherapy in neonatal jaudice.
Related Topics
