Removal of Nitrogen From Amino Acids
| Home | | Biochemistry |Chapter: Biochemistry : Amino Acids: Disposal of Nitrogen
The presence of the α-amino group keeps amino acids safely locked away from oxidative breakdown. Removing the α-amino group is essential for producing energy from any amino acid and is an obligatory step in the catabolism of all amino acids.
REMOVAL OF NITROGEN FROM AMINO ACIDS
The presence of the
α-amino group keeps amino acids safely locked away from oxidative breakdown.
Removing the α-amino group is essential for producing energy from any amino
acid and is an obligatory step in the catabolism of all amino acids. Once
removed, this nitrogen can be incorporated into other compounds or excreted as
urea, with the carbon skeletons being metabolized. This section describes
transamination and oxidative deamination, reactions that ultimately provide
ammonia and aspartate, the two sources of urea nitrogen.
A. Transamination: the funneling of amino groups to glutamate
The first step in the
catabolism of most amino acids is the transfer of their α-amino group to
α-ketoglutarate (Figure 19.7), producing an α-keto acid (derived from the
original amino acid) and glutamate. α-Ketoglutarate plays a pivotal role in
amino acid metabolism by accepting the amino groups from most amino acids,
thereby becoming glutamate. Glutamate produced by transamination can be
oxidatively deaminated (see below) or used as an amino group donor in the
synthesis of nonessential amino acids. This transfer of amino groups from one
carbon skeleton to another is catalyzed by a family of enzymes called
aminotransferases (also called transaminases). These enzymes are found in the
cytosol and mitochondria of cells throughout the body. All amino acids, with
the exception of lysine and threonine, participate in transamination at some
point in their catabolism. [Note: These two amino acids lose their α-amino
groups by deamination.]
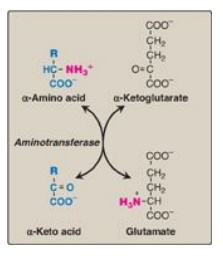
Figure 19.7 Aminotransferase reaction using α-ketoglutarate as the amino group acceptor.
1. Substrate specificity of aminotransferases: Each aminotransferase is specific for one or, at most, a few amino group donors. Aminotransferases are named after the specific amino group donor, because the acceptor of the amino group is almost always α-ketoglutarate. Two important aminotransferase reactions are catalyzed by alanine aminotransferase (ALT) and aspartate aminotransferase (AST), as shown in Figure 19.8).
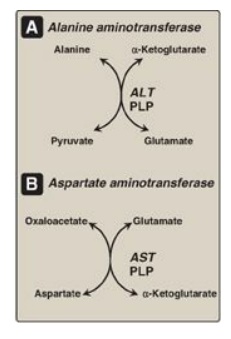
Figure 19.8 Reactions catalyzed during amino acid catabolism. A. Alanine aminotransferase (ALT). B. Aspartate aminotransferase (AST). PLP = pyridoxal phosphate.
a. Alanine aminotransferase: ALT is present in many tissues. The
enzyme catalyzes the transfer of the amino group of alanine to α-ketoglutarate,
resulting in the formation of pyruvate and glutamate. The reaction is readily
reversible. However, during amino acid catabolism, this enzyme (like most
aminotransferases) functions in the direction of glutamate synthesis. [Note:
Glutamate, in effect, acts as a “collector” of nitrogen from most amino acids.]
b. Aspartate aminotransferase: AST is an exception to the rule
that aminotransferases funnel amino groups to form glutamate. During amino acid
catabolism, AST transfers amino groups from glutamate to oxaloacetate, forming
aspartate, which is used as a source of nitrogen in the urea cycle. Like other
transaminations, the AST reaction is reversible.
2. Mechanism of action of aminotransferases: All aminotransferases require the
coenzyme pyridoxal phosphate (a derivative of vitamin B6), which is covalently
linked to the ε-amino group of a specific lysine residue at the active site of
the enzyme. Aminotransferases act by transferring the amino group of an amino
acid to the pyridoxal part of the coenzyme to generate pyridoxamine phosphate.
The pyridoxamine form of the coenzyme then reacts with an α-keto acid to form
an amino acid, at the same time regenerating the original aldehyde form of the
coenzyme. Figure 19.9 shows these two component reactions for the reaction
catalyzed by AST.
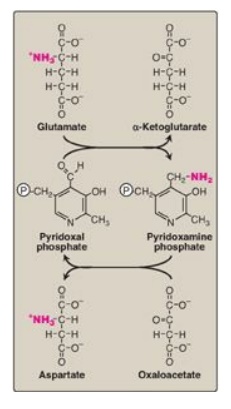
Figure 19.9 Cyclic
interconversion of pyridoxal phosphate and pyridoxamine phosphate during the
aspartate aminotransferase reaction.P
= phosphate group.
3. Equilibrium of transamination reactions: For most transamination reactions,
the equilibrium constant is near 1. This allows the reaction to function in
both amino acid degradation through removal of α-amino groups (for example,
after consumption of a protein-rich meal) and biosynthesis of nonessential
amino acids through addition of amino groups to the carbon skeletons of α-keto
acids (for example, when the supply of amino acids from the diet is not
adequate to meet the synthetic needs of cells).
4. Diagnostic value of plasma aminotransferases: Aminotrans-ferases are normally
intracellular enzymes, with the low levels found in the plasma representing the
release of cellular contents during normal cell turnover. Elevated plasma
levels of aminotransferases indicate damage to cells rich in these enzymes. For
example, physical trauma or a disease process can cause cell lysis, resulting
in release of intracellular enzymes into the blood. Two aminotransferases, AST
and ALT, are of particular diagnostic value when they are found in the plasma.
a. Liver disease: Plasma AST and ALT are elevated in nearly all
liver diseases but are particularly high in conditions that cause extensive
cell necrosis, such as severe viral hepatitis, toxic injury, and prolonged
circulatory collapse. ALT is more specific than AST for liver disease, but the
latter is more sensitive because the liver contains larger amounts of AST.
Serial measurements of AST and ALT (so-called “liver function tests”) are often
useful in determining the course of liver damage. Figure 19.10 shows the early
release of ALT into the serum, following ingestion of a liver toxin. [Note:
Elevated serum bilirubin results from hepatocellular damage that decreases the
hepatic conjugation and excretion of bilirubin.]
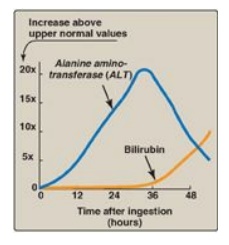
Figure 19.10 Pattern of serum
ALT and bilirubin in the plasma, following poisoning with the toxic mushroom
Amanita phalloides.
b. Nonhepatic disease: Aminotransferases may be elevated in nonhepatic diseases such as those that cause damage to cardiac or skeletal muscle. However, these disorders can usually be distinguished clinically from liver disease.
B. Oxidative deamination of amino acids
In contrast to
transamination reactions that transfer amino groups, oxidative deamination
reactions result in the liberation of the amino group as free ammonia (Figure
19.11). These reactions occur primarily in the liver and kidney. They provide
α-keto acids that can enter the central pathways of energy metabolism and
ammonia, which is a source of nitrogen in hepatic urea synthesis. [Note:
Ammonia exists primarily as ammonium (NH4+) in aqueous
solution, but it is the un-ionized form (NH3) that crosses
membranes.]
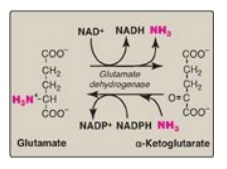
Figure 19.11 Oxidative deamination by glutamate dehydrogenase. [Note: The enzyme is unusual in that it uses both NAD+ (nicotinamide adenine dinucleotide) and NADPH (nicotinamide adenine dinucleotide phosphate).]
1. Glutamate dehydrogenase: As described above, the amino
groups of most amino acids are ultimately funneled to glutamate by means of
transamination with α-ketoglutarate. Glutamate is unique in that it is the only
amino acid that undergoes rapid oxidative deamination, a reaction catalyzed by
glutamate dehydrogenase (see Figure 19.11). Therefore, the sequential action of
transamination (resulting in the transfer of amino groups from most amino acids
to α-ketoglutarate to produce glutamate) and the oxidative deamination of that
glutamate (regenerating α-ketoglutarate) provide a pathway whereby the amino
groups of most amino acids can be released as ammonia.
a. Coenzymes: Glutamate dehydrogenase, a mitochondrial enzyme, is unusual in that it can use either nicotinamide adenine dinucleotide (NAD+) or its phosphorylated reduced form (NADPH) as a coenzyme (see Figure 19.11). NAD+ is used primarily in oxidative deamination (the simultaneous loss of ammonia coupled with the oxidation of the carbon skeleton, as shown in Figure 19.12A), and NADPH is used in reductive amination (the simultaneous gain of ammonia coupled with the reduction of the carbon skeleton, as shown in Figure 19.12B).
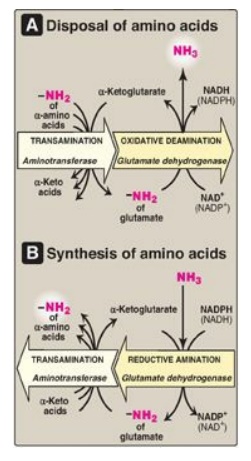
Figure 19.12 Combined actions of aminotransferase and glutamate dehydrogenase reactions. [Note: Reductive amination occurs only when ammonia (NH3) level is high.] NAD(H) = nicotinamide adenine dinucleotide; NADP(H) = nicotinamide adenine dinucleotide phosphate.
b. Direction of reactions: The direction of the reaction
depends on the relative concentrations of glutamate, α-ketoglutarate, and
ammonia and the ratio of oxidized to reduced coenzymes. For example, after
ingestion of a meal containing protein, glutamate levels in the liver are
elevated, and the reaction proceeds in the direction of amino acid degradation
and the formation of ammonia (see Figure 19.12A). High ammonia levels are
required to drive the reaction to glutamate synthesis.
c. Allosteric regulators: Guanosine triphosphate is an allosteric inhibitor of glutamate dehydrogenase, whereas adenosine diphosphate (ADP) is an activator. Therefore, when energy levels are low in the cell, amino acid degradation by glutamate dehydrogenase is high, facilitating energy production from the carbon skeletons derived from amino acids.
2. D-Amino acid oxidase: D-Amino acids are found in plants
and in the cell walls of microorganisms but are not used in the synthesis of
mammalian proteins. D-Amino acids are, however, present in the diet and are
efficiently metabolized by the kidney and liver. D-Amino acid oxidase (DAO) is
a flavin adenine dinucleotide– dependent peroxisomal enzyme that catalyzes the
oxidative deamination of these amino acid isomers, thereby producing α-keto
acids, ammonia, and hydrogen peroxide. The α-keto acids can enter the general
pathways of amino acid metabolism and be reaminated to L-isomers or catabolized
for energy. [Note: DAO degrades D-serine, the isomeric form of serine that
modulates N-methyl-D-aspartate (NMDA)-type glutamate receptors. Increased DAO
activity has been linked to increased susceptibility to schizophrenia.] L-amino
acid oxidases are known, but their physiologic significance is unclear.
C. Transport of ammonia to the liver
Two mechanisms are
available in humans for the transport of ammonia from the peripheral tissues to
the liver for its ultimate conversion to urea. Both are important in, but not
exclusive to, skeletal muscle. The first uses glutamine synthetase to combine
ammonia with glutamate to form glutamine, a nontoxic transport form of ammonia
(Figure 19.13). The glutamine is transported in the blood to the liver where it
is cleaved by glutaminase to produce glutamate and free ammonia. The ammonia is
converted to urea. The second transport mechanism involves the formation of
alanine by the transamination of pyruvate produced from both aerobic glycolysis
and metabolism of the succinyl coenzyme A (CoA) generated by the catabolism of
the branched-chain amino acids isoleucine and valine. Alanine is transported by
the blood to the liver, where it is converted to pyruvate, again by
transamination. The pyruvate is used to synthesize glucose, which can enter the
blood and be used by muscle, a pathway called the glucose–alanine cycle.
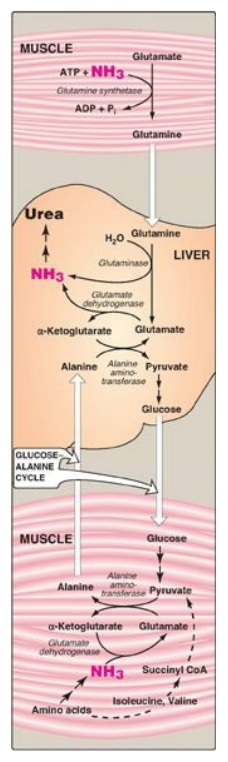
Figure 19.13 Transport of ammonia (NH3) from muscle to the liver. ADP = adenosine diphosphate; Pi = inorganic phosphate; CoA = coenzyme A.
Related Topics
