Combination and other strategies for colon targeting
| Home | | Pharmaceutical Drugs and Dosage | | Pharmaceutical Industrial Management |Chapter: Pharmaceutical Drugs and Dosage: Organ-specific drug delivery
Physiological differences between the colon and the small intestine, such as intraluminal pressure and the level of hydration, can also be utilized to design a colon-targeted DDS.
Combination and
other strategies for colon targeting
Physiological
differences between the colon and the small intestine, such as intraluminal pressure
and the level of hydration, can also be utilized to design a colon-targeted
DDS. For example, Takada and colleagues uti-lized the higher intraluminal
pressure in the colon and its low hydration state as a trigger mechanism for
drug release. To utilize this as a trigger for drug release, the authors
prepared liquid-filled hard gelatin capsules coated with an insoluble ethyl
cellulose film. The drug was dissolved in a water soluble or insoluble
semisolid base, such as PEG 1000, which liquifies at body temperature. After
oral administration of the capsule, it behaves as a flexible membrane balloon
with encapsulated drug, thus maintaining integrity during small intestinal
transit. On reaching the colon, reabsorption of water leads to increased viscosity
of the contents of the ethyl cellulose balloon, leading to its fragility and
disintegration under higher pressure. The authors identified the thickness of
the water-insoluble ethyl cellulose membrane as the key factor that controls
drug release. Using this system, the authors demonstrated targeted delivery to
the human colon using caffeine as a model drug and glycyrrhizin in dogs.
In
addition to targeted drug release in the colon, the dosage form may incorporate
a bioadhesive polymer to prolong the duration of time the dos-age form stays in
the colon. The polymers that can be used for this purpose include
polycarbophils, polyurethanes, and poly(ethylene oxide—propylene oxide)
copolymers. Utilizing this strategy, Kakoulides et al. synthesized azo cross-linked
bioadhesive acrylic polymers. The cross-linking prevents hydra-tion and
swelling in the upper intestinal tract. On degradation of azo bonds in the
large intestine, hydrogel swelling and bioadhesion was expected to lead to drug
release and prolonged residence in the colonic environment.
Similarly,
Gao et al. synthesized a conjugate of bioadhesive polymer
N-(2-hydroxypropyl)methacrylamide (HPMA) and the drug 9-aminocamp-tothecin
(9-AC) via a spacer containing a combination of an aromatic azo bond and a 4-aminobenzylcarbamate
group. The spacer was designed to release the drug by azo bond cleavage in the
colonic microenvironment. In subsequent studies, the authors observed colon
targeting in mouse53 and rat54 models for the treatment of colon cancer. After
oral administration of equal doses of the polymer conjugate or free 9-AC to
mice, colon-specific release of 9-AC produced high local concentrations with
the mean peak concentra-tion of 9-AC in cecal contents, feces, cecal tissue,
and colon tissue being 3.2, 3.5, 2.2, and 1.6-fold higher, respectively.
Therefore, the authors anticipated higher antitumor efficacy of the polymer
conjugate due to prolonged colon tumor exposure to higher and more localized
drug concentrations.
Combination
strategies for colon-specific drug delivery commonly utilize a combination of
pH and colonic microflora-based strategies. For example, Kaur and Kim prepared
prednisolone beads with multiple coating layers for colonic delivery of the
anti-inflammatory compound. The authors coated prednisolone on nonpareil beads
followed by a hydrophobic coat of Eudragit® RL/RS; followed by a layer
containing chitosan, suc-cinic acid, and Eudragit® RL/RS; followed by an
outermost enteric coat layer (Figure 15.7a). In vitro experiments showed absence of
drug release in simulated gastric and intestinal fluids, followed by drug
release in the
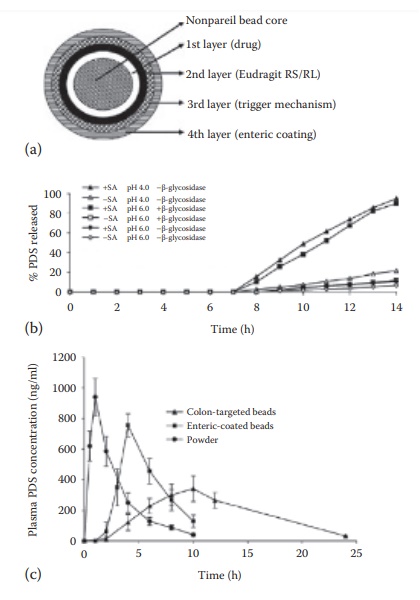
Figure 15.7 Colonic drug targeting. Combination strategy for colonic drug targeting
using an oral solid dosage form: (a) design of the targeted drug delivery
system. Predniosolone (PDS, drug) was coated on nonpareil beads (1st layer),
followed by a hydrophobic coat of Eudragit® RS/RL polymers (2nd layer), which
was fol-lowed by a layer of Eudragit® RS/RL polymers in combination with chitosan
and succinic acid (3rd layer), and the outermost enteric coating layer of
Eudragit® L 100 (4th layer), (b) In vitro drug release from the system as a
function of pH, succinic acid (SA) content in the formulation, and
β-glucosidase content in the dissolution medium. The formulation dissolution
was carried out in the gastric fluid for the first 2 h, followed by the small
intestinal fluid for next 5 h, and the pathological colonic fluid for the last
7 h, and (c) plasma drug concentration after oral administration of powder,
enteric coated, or colon targeted drug delivery systems in rats. (Modified from
Kaur, K., Kim, K., Int. J. Pharm., 366, 140, 2009. With Permission.)
pathological
colonic fluid with rate dependence on the presence of suc-cinic acid in the formulation
and the presence of the enzyme β-glucosidase
(Figure 15.7b). The authors proposed a
combination mechanism of drug release that involved pH-triggered enteric
dissolution of the outer-most layer, followed by chitosan and Eudragit®
swelling in the presence of succinic acid, and biodegradation of chitosan by
the colonic bacteria. Organic acid interacts with the amine groups in Eudragit®
and chitosan polymers, leading to increased permeability of the coating and
facilitated drug release at the colonic site. On oral administration of this formulation
to male Sprague-Dawley rats, significant delay in the time to maximum plasma
drug concentration (Tmax)
was obtained compared to both unmodi-fied powder and enteric-coated tablet
formulations, thus indicating colonic targeting (Figure
15.7c).
Kidney-targeted drug delivery
Kidney-targeted
drug delivery is quite promising to improve drug efficacy and safety in the
treatment of renal diseases.1 Renal targeting
is valuable to avoid extrarenal side effects of drugs used in the treatment of
kidney diseases or to optimize the intrarenal distribution of a drug candidate,
thus increasing its therapeutic index. Although renal drug delivery is not well
studied, it highlights the challenges and opportunities inherent in develop-ing
a targeted DDS. Among the drugs used for the treatment of kidney diseases are
anti-inflammatory and antifibrotic compounds. Specific drug delivery to the
kidney may also be helpful during shock, renal transplanta-tion, ureteral
obstruction, diabetes, renal carcinoma, and other diseases such as Fanconi and
Bartter’s syndrome. In addition, renal targeting can be helpful for drugs that
would otherwise be rapidly metabolized and inac-tivated before reaching the
kidney and to overcome or minimize the effects of pathological conditions, such
as proteinuria, on drug distribution to the target site.
Cellular drug targets
Three
cellular drug targets have been identified within the kidney— (1) proximal
tubular cells, (2) mesangial cells, and (3) fibroblasts. Nephron, the
functional unit of the kidney, consists of a renal corpuscle and a renal
tubule. The renal corpuscle is responsible for blood filtration. It consists of
the glomerulus and the Bowman’s capsule. The renal tubule consists of proximal
and distal convoluted tubules interconnected by the loop of Henle. After blood
filtration through the glomerulus, the proximal con-voluted tubule is
responsible for pH regulation and reabsorption of salts and organic solutes
from the filtrate. The luminal surface of the proximal tubular cells has a
brush-border epithelium, with densely packed microvilli, which help increase
the luminal surface area.
Mesangium,
or the mesangial tissue, constitutes the inner layer of glom-erulus, within the
basement membrane of the renal corpuscle. It surrounds the glomerular arteries
and arterioles both within (intraglomerular) or outside (extraglomerular) the
glomerulus. The glomerular epithelium is fenestrated and there is no basement
membrane between the glomerular capillaries and the mesangial cells. Hence,
mesangial cells are separated from the capillary lumen by only a layer of
endothelial cells. Mesangial cells are phagocytic in nature and secrete an
amorphous, basement mem-brane-like material, known as the mesangial matrix.
These cells generate inflammatory cytokines and are involved in the uptake of
macromolecules.
Fibroblasts
synthesize ECM and collagen. Excessive production and accumulation of the ECM
lead to fibrosis. Renal fibrosis is the underlying process that leads to the
progression of chronic kidney disease to end-stage renal disease. It involves
changes in the renal vasculature, glomeruloscle-rosis, and tubulointerstitial
fibrosis. Of these, tubulointerstitial fibrosis is considered to be the most
consistent predictor of an irreversible loss of renal function and progression
to end-stage renal disease. The accumulation of ECM components in fibrotic
disease is attributed to the activation of resi-dent interstitial fibroblasts.
Therefore, targeted drug delivery to renal fibro-blasts has been attempted. For
example, Kushibiki et al. used cationized gelatin to complex an enhanced green
fluorescent protein (EGFP) express-ing plasmid, which was injected into the
left kidney of mice through the ureter. The authors observed significant EGFP
expression in the fibroblasts residing in the renal interstitial cortex.
Similarly, Xia et al. reported the delivery of siRNA targeted against heat
shock protein 47 (HSP47) using cationized gelatin microspheres to the mice
kidneys with tubulointersti-tial fibrosis. The authors observed that the
cationized gelatin microspheres enhanced and prolonged the antifibrotic effect
of the siRNA.
Of
these cell types, the proximal tubular cells have been the target of most
drug-delivery strategies. They are metabolically the most active cells in the
kidney and are involved with the transport and metabolism62 of several organic
and inorganic substrates. Consequently, they have spe-cific transporter
receptors on their luminal and basolateral membranes for substrate exchange
between the blood and the urine. These transport and metabolic functions of the
proximal tubular epithelial cells are utilized for drug targeting.
Particulate systems
The
lack of basement membrane in the glomerular capillaries makes mesangial cells
in close contact with the bloodstream, being separated from the capillary lumen
by only a layer of endothelial cells. The mesangial cells, therefore, can be
targeted using particulate carrier systems that may not filter through the
glomeruli. Tuffin et al. used OX7-coupled immu-noliposomes to target renal
mesangial cells. The authors coupled OX7 monoclonal antibody F(ab′)2 fragments, directed
against the mesangial cell expressing Thy1.1 antigen, on the surface of
doxorubicin-loaded immu-noliposomes. The authors observed specific targeting to
rat mesangial cells in vitro and in vivo on intravenous administration.
Administration of doxorubicin-encapsulated immunoliposomes resulted in
significant glo-merular damage, with low damage to other parts of the kidney
and other organs. The targeted localization was not observed with free drug or
lipo-somes, and immunoliposome localization was blocked by coadministration of
free antibody fragments.
In
a later study, the authors attempted to correlate the biodistribution of these
immunoliposomes with the tissue distribution of the antigen. The Thy1.1 antigen
showed high expression in rat glomeruli, brain cortex and striatum, and thymus;
and moderate expression in the collecting ducts of the kidney, lung, and
spleen. The biodistribution of immunoliposomes did not correlate well with the
tissue distribution of Thy1.1 antigen, with the highest levels seen in the
spleen, followed by lungs, liver, and kidney. Within the kidney, specific
localized delivery to the mesangial cells was observed, which was sensitive to
competition with the unbound OX7 monoclonal antibody fragments. The authors concluded
that the absence of endothelial barriers and high target antigen density are
important factors governing tissue localization of immunoliposomes.
An
application of drug targeting to glomerular endothelial cells to reduce
systemic side effects of drug therapy was reported by Asgeirsdottir et al., who
used immunoliposomes to target glomerular endothelial cells in mice.
Glomerulonephritis, a spectrum of inflammatory diseases specifically affecting
renal glomeruli, is characterized by the activa-tion of proinflammatory
pathways, resulting in glomerular injury and proteinuria. These disorders are
frequently treated with glucocorticoids, such as dexamethasone, in combination
with cytotoxic agents, such as cyclophosphamide, as anti-inflammatory and
immunosuppressive agents. These drugs, however, present serious extrarenal side
effects including an increase in blood glucose levels with dexamethasone.
Asgeirsdottir et al. coupled monoclonal rat anti-mouse E-selectin antibody,
MES-1, to the surface of liposomes. The selection of this antibody was designed
to tar-get glomerular endothelial cells in glomerulonephritis, wherein
endothelial cell expression of inflammation-related cell-adhesion molecules,
such as E-selectin and VCAM-1, is upregulated. The authors obtained
site-specific delivery of immunoliposome encapsulated anti-inflammatory agent
dexa-methasone and observed reduction in glomerular proinflammatory gene
expression with no effect on blood glucose levels.
In
addition to liposomes, nanoparticles have been utilized for drug targeting to
the mesangial cells. For example, Manil et al. used isobutyl-cyanoacrylate
nanoparticles for targeting the antibiotic actinomycin D to rat mesangial
cells. Compared to the free drug, the uptake of drug-loaded nanoparticles in
the whole kidneys was over two fold at both 30 and 120 min after intravenous
injection. Similar or higher uptake ratios were obtained for isolated rat
glomeruli, but not for tubules. The glomerular uptake of nanoparticles was even
higher in rats with experimental glomerulonephritis. Mesangial cell targeting
was indicated by in vitro
experiments, which dem-onstrated fivefold higher uptake by mesangial cells than
the epithelial cells. In a separate study, Guzman et al. also obtained about
twofold higher in vitro uptake of
drug-loaded nanoparticles in rat mesangial cells using polycapro-lactone as the
polymeric carrier and digitoxin as the drug candidate.
The prodrug approach
Prodrugs
are drug conjugates designed to modify the physicochemical and/or biopharmaceutical
properties of the drug candidate. Their derivatization is bioreversible and is
designed to improve drug properties with respect to solubility, stability,
permeability, presystemic metabolism, and targeting. Prodrugs retain the advantages of low molecular weight compounds such as low
immunogenicity and feasibility of oral administration. Renal speci-ficity of
prodrugs would depend on the renal-specific metabolism and/or uptake of the
promoiety. For this purpose, amino acid prodrugs, which can be activated by
kidney-specific enzymes, have been evaluated for renal targeting.
Amino
acid prodrugs have advantages of biodegradability in addition to
receptor-mediated uptake, which can help in both oral absorption and organ or
tissue-specific targeting. For example, valine prodrugs of acyclo-vir and
ganciclovir showed 3–5 times higher bioavailability than the par-ent compounds. Enhanced oral
absorption of amino acid prodrugs is attributed to carrier-mediated intestinal
uptake via transporters. For organ and tissue-specific drug targeting, the
l-glutamate transport system has been commonly utilized.
Prodrug
design for renal targeting is aimed at utilizing the kidney-specific enzymes.
The proximal tubular cells contain high levels of metabolizing enzymes in the
cytosol (such as l-amino acid decarboxylase, β-lyase, and N-acetyl transferase) and at the brush border (such as γ-glutamyl transpep-tidase). Examples
of renal-targeted prodrugs include the γ-glutamyl pro-drugs of l-dopa and
sulfamethoxazole.
Gludopa
(γ-l-glutamyl-l-dopa)
is a kidney-specific dopamine prodrug. Cummings et al. reported its
pharmacokinetic and tissue distribution in rats. Gludopa was metabolized
primarily in the liver and kidney, with dopamine being the major kidney
metabolite. The pharmacokinetics of gludopa in healthy human volunteers
indicated urinary dopamine excre-tion in parallel with urinary levodopa
excretion, supporting the view that levodopa was the precursor of urinary
dopamine. Based on these results, Boateng et al. indicated that gludopa may be
useful in conditions where renal effects of dopamine are indicated. However,
Lee noted the limitations in clinical practice posed by its low oral
bioavailability in humans.
Kidney-specific
delivery of parent compounds after IV administration of γ-l-glutamyl (G) and N-acetyl-γ-l-glutamyl (AG) prodrugs of
p-nitroaniline, sulfamethoxazole,
and sulphamethizole was investigated by Murakami et al. in rats. The authors
observed higher plasma stability of AG over G prodrugs for all compounds. The
concentration of parent compounds was higher in the kidney than the pulmonary
and hepatic tissue for all compounds, with markedly increased kidney
distribution of AG prodrugs of p-nitroaniline and sulfamethoxazole. The
activation of AG prodrugs requires the action of two enzymes—deacylation by N-acylamino acid deacylase and
hydrolysis by γ-glutamyl
transpeptidase, whereas G prodrugs can be activated by the action of γ-glutamyl transpeptidase alone. When
biodistribution of G pro-drugs of sulfamethoxazole was studied in mice,
relatively high concentra-tions of sulfamethoxazole were found in nonrenal
tissues as well indicating rapid kinetics of enzymatic cleavage of G prodrugs
even in tissues with low γ-glutamyl
transpeptidase activity. However,
kidney selective accumula-tion was obtained after the administration of AG
prodrugs. Drieman et al. hypothesized that the renal selectivity of AG prodrugs
of sulfamethoxazole was due to a carrier-mediated transport followed by
intracellular conversion of the prodrug to the active compound.
Effective
utilization of the prodrug strategy requires intensive investiga-tion of the
role of variables such as the linker groups and the promoiety modifications.
This results in an inherent complexity in prodrug design and utilization for
organ or tissue-targeted drug delivery.
Bioconjugation approaches
Bioconjugation
of a drug to a carrier that is significantly larger than the molecular size of
the drug allows the biopharmaceutical properties of car-rier to dominate the
absorption and biodistribution of the conjugate. In the case of renal drug
targeting to the proximal tubular cells, the conjugates would need to be
filtered through the glomerular capillaries and reabsorbed by the tubular
cells. Particles with a hydrodynamic diameter below 5–7 μm are rapidly filtered through the
glomerulus.
For
this purpose, the carriers that naturally accumulate in the prox-imal tubular
cells can be used as drug carriers. These include the low (less than about 30
kDa) molecular weight proteins (LMWPs), such as lysozyme, aprotinin, and
cytochrome C. They are readily filtered through the glomerulus but selectively
reabsorbed by the proximal tubular cells (Figure 15.8a).
Thus, LMWP–drug conjugates that are stable in the plasma but cleaved within the
proximal tubular cells after endosomal/lyso-somal uptake can be used as
effective vehicles for drug targeting. Drugs may be conjugated to LMWPs
directly using the lysine amino groups or with a spacer. Relatively large size
of the LMWPs allows the pharmaco-kinetic properties of the LMWPs to override
those of the drug candidate in the LMWP–drug conjugates. This approach,
however, is limited by the
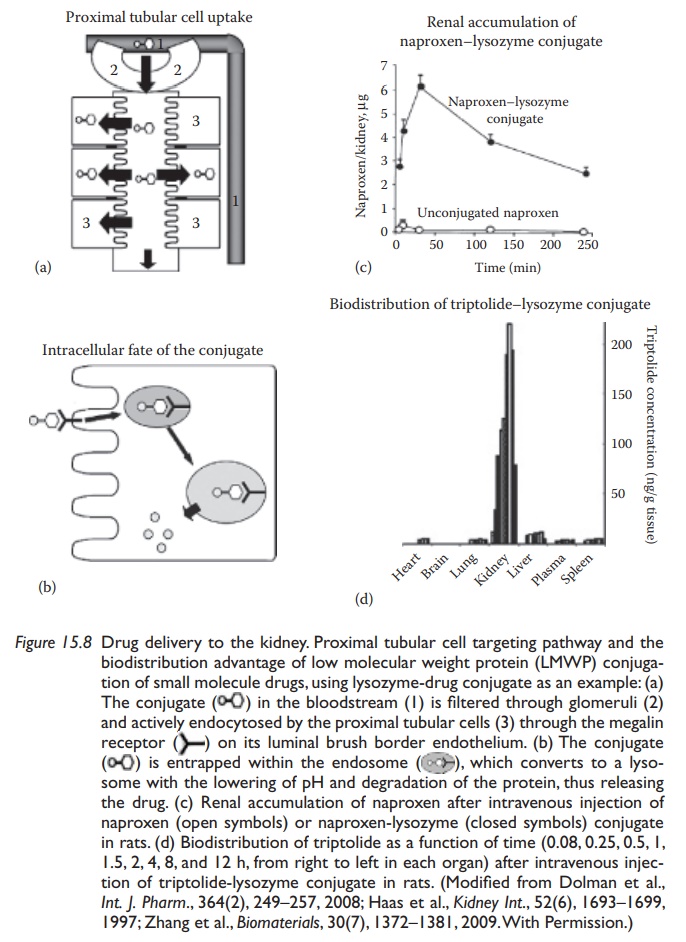
The
internalization of proteins in the proximal convoluted tubule epithe-lium cells
is mediated via the multiligand megalin and cubilin receptors. These cells have
very high endocytic activity. After endocytosis, the protein is degraded in the
lysosomes, wherein the attached drug may be released (Figure
15.8b). Lysozyme has been used as a renal carrier for the non-steroidal
anti-inflammatory drug (NSAID) naproxen (Figure 15.8c),
the acetylcholinesterase (ACE) inhibitor compound captoril, and the
nephroprotective compound triptolide (Figure 15.8d).
Naproxen
is a carboxylic acid group-bearing compound that could be conjugated to the amine
group in the lysozyme directly by an amide bond or through lactic acid spacer
by an ester bond. The biodistribution and degradation of these conjugates was
compared with lysozyme and naproxen by themselves in rats. Drug conjugation did
not affect the renal uptake or degradation of lysozyme in the rat kidney. The
pharmacokinetic profile of the conjugates was similar to that of lysozyme, but
markedly different from the drug. The drug was rapidly taken up by and degraded
in the kid-ney with no detectable levels in the plasma (Figure 15.8c). Similar results were obtained when captopril was
conjugated with lysozyme through a spacer utilizing disulfide linkage.
Targeting this ACE inhibitor to the kid-ney was hypothesized to prevent
attenuation of renoprotective (antiprotein-uric) efficacy of captopril under
high sodium concentrations. The drug was efficiently targeted to the kidney
with the rapid release of the drug.
Triptolide
is an immunosuppressive and anti-inflammatory natural compound with low water
solubility and significant toxicity. Renal target-ing of triptolide–lysozyme
conjugate linked through succinyl residue was investigated in rats. The authors
obtained significantly higher targeting efficiency of the drug conjugate to the
kidney with reversal of disease pro-gression in renal ischemia-reperfusion
injury rat model, lower hepatotox-icity, and no effect on immune and genital
systems, compared to the free drug (Figure 15.8d).
These results demonstrated the potential therapeutic benefits of renal drug targeting.
In
addition to the use of LMWPs as drug targeting ligands, their receptor-mediated
uptake can also be utilized to mitigate renal toxicity of drugs. For example,
endocytosis by proximal tubular cells is responsible for the renal accumulation
and toxicity of aminoglycoside antibiotics, such as gentami-cin, which is a
substrate of the megalin receptors. Watanabe et al. found that coadministration
of cytochrome C competes with receptor-mediated renal uptake of gentamicin,
thus reducing its renal accumulation in rats. However, the required dose of cytochrome C was quite high; the authors tested
the relative efficacy of peptide fragments in reducing the renal accu-mulation
of gentamicin. Three peptide fragments derived from actin-regulating proteins
were identified that reduced the renal accumulation of gentamicin without
affecting its plasma concentration-time profile.
In
addition to the exploitation of LMWPs for modulating the pharma-cokinetics and
biodistribution of drugs by utilizing their physiological disposition to modify
drug biodistribution, drugs and enzymes can also be targeted to the renal
proximal tubular epithelial cells by their surface modification. For example,
Inoue et al. modified the enzyme superoxide dismutase (SOD), which disproportionates
the superoxide free radical into oxygen and hydrogen peroxide—thus reducing
free radical and oxidative stress in the cells. Intravenously administered Cu,
Zn–SOD is rapidly removed from the circulation with a half-life of about 5 min
and appears intact in the urine, thus indicating that it is filtered through
the glomeru-lus. The authors conjugated hexamethylene diamine (AH) to SOD. The
conjugate (AH–SOD) was rapidly filtered through the glomeruli but bound apical
plasma membranes of proximal tubular cells followed by localized action in
these cells. The authors observed more than 80% of the radio-activity derived
from AH–SOD localized in the kidney at 30 min after injection, most of which
was localized in the proximal tubular cells. In vitro kinetic studies
revealed that the specific binding of AH–SOD to apical surface of the tubular cells was attributable to AH.
Polymeric
carriers have also been described for renal drug targeting. These include the
anionized derivatives of polyvinylpyrrolidone (PVP), low molecular weight N-(hydroxypropyl) methylacrylamide
(HPMA), and low molecular weight chitosan. The use of synthetic polymers
requires surface modification and derivatizing groups for optimum renal
accumula-tion. For example, PVP by itself does not accumulate in the tubular
epithe-lial cells, but on copolymerization with maleic acid, it selectively
distributed into the kidneys on IV injection in mice. When anionized
derivatives of PVP were prepared, the plasma clearance of these derivatives
decreased with increasing size of anionic groups. In addition, even though the
clear-ance of carboxylated PVP and sulfonated PVP from the blood was similar,
renal accumulation of carboxylated PVP was several fold higher than that of
sulfonated PVP. In summary, these studies demonstrate not only the potential
for renal targeting of drugs where it may be beneficial but also the potential
to prevent accumulation in the kidney for drugs that have renal toxicity.
Related Topics
