Colon-targeted drug delivery
| Home | | Pharmaceutical Drugs and Dosage | | Pharmaceutical Industrial Management |Chapter: Pharmaceutical Drugs and Dosage: Organ-specific drug delivery
Traditionally, colonic drug delivery is focused on the treatment of local conditions such as ulcerative colitis, colorectal cancer, irritable bowel syndrome, amebiasis, and Crohn’s disease.
Colon-targeted
drug delivery
Traditionally,
colonic drug delivery is focused on the treatment of local conditions such as
ulcerative colitis, colorectal cancer, irritable bowel syndrome, amebiasis, and
Crohn’s disease. However, it has been gaining importance for the systemic
delivery of potent compounds such as proteins, peptides, and oligonucleotides
that are unstable in the harsh conditions of the upper GI tract. As colon is
rich in lymphoid tissues, it offers opportuni-ties for the oral delivery of
vaccines targeted for release and absorption in the lower GI tract. In
addition, colon delivery can be exploited to improve the bioavailability of
drugs that are extensively metabolized by cytochrome P450 enzymes in the upper
GI tract, because the activity of these metabo-lizing enzymes are relatively
lower in the colonic mucosa. Colon-specific drug delivery may also help
overcome GI side effects of drugs. For example, conversion of flurbiprofen to a
glycine prodrug, hydrolysable by colonic microfloral enzymes (amidases),
reduced its ulcerogenic activity in rats. Targeted drug delivery to the colon has been extensively studied.
Colon-specific
drug delivery is challenged by its distal location in the GI tract. Even
localized delivery through the rectum, however, only reaches a small part of
the colon and is not a patient-friendly mode of adminis-tration. Therefore,
oral delivery has been explored, utilizing physiological differences in the
colonic microenvironment and physiology. The aspects of colon physiology that
have been exploited to develop drug-targeting strategies include the presence
of unique colonic microflora, high pH, the relatively predictable transition
time in the small intestine, and high intra-luminal pressure inside the colon.
In addition, osmotically and oxidation potential controlled DDSs, and
bioadhesive polymers have been used for colonic drug delivery.
Utilization of the unique colonic microflora
Human
colonic microflora consists predominantly of bacteria, which also make up to 60%
of the dry mass of feces. The metabolic activities of this microflora results
in the salvage of absorbable nutrients from diet by fermenting unused energy
substrates, trophic effects on the epi-thelium, and protection of the colonized
host against invasion by alien microbes. Colonic bacteria are mostly gram
negative and anaerobic, except cecum, which can have high amount of aerobic
bacteria. Bacteria in the proximal part of the colon are primarily involved in
ferment-ing carbohydrates, whereas the latter part breaks down proteins and
amino acids.
The
unique metabolic ability of these microbes has been exploited to develop
polymerics and prodrugs that are degraded by the unique enzy-matic activities
of colonic microflora. In particular, the azo reductase and glycosidase
activities of the microflora help degrade the azo bound and glycosidic
linkages. Prodrug strategy for colonic drug delivery utilizes drug conjugation
with a promoiety through an azo bond, which is degraded by the colonic
bacteria. Examples of such prodrugs include sulfasalazine, balsalazide,
ipsalazide, olsalazide, and salicylazosulfapyridine for the treatment of
inflammatory bowel disease. As shown in Figure 15.5,
these
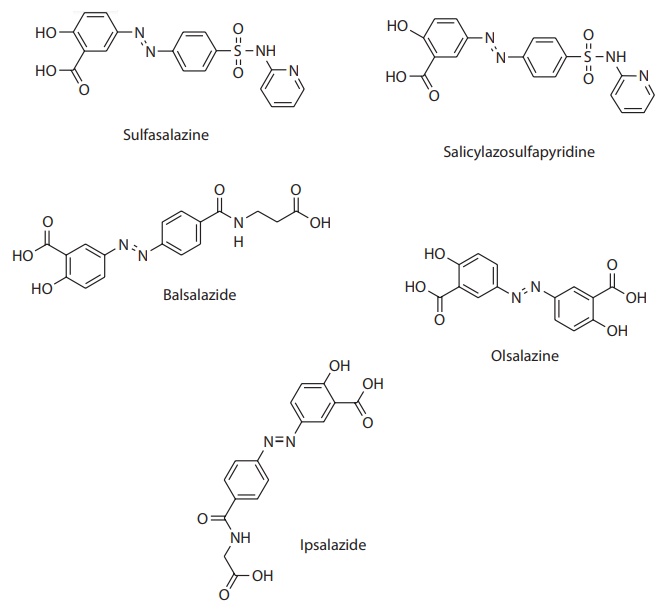
Figure 15.5 Colon-targeted drug delivery by prodrug strategy. Prodrug strategy for
colonic drug delivery utilizes drug conjugation with a promoiety through an azo
bond, which is reductively cleaved by the colonic anaerobic bacteria to release
the parent compound. This figure shows the structure of several prodrugs of
5-amino salicylic acid (5-ASA), an anti-inflammatory compound used for the
treatment of inflammatory bowel disease.
Sulfasalazine was first introduced for the treatment of rheumatoid
arthritis and inflammatory bowel disease. In the colon, it degrades into 5-ASA
and sulfapyridine, which is responsible for most of the side effects of
sulfasalazine. This problem was overcome by the use of other promoieties, such
as 4-amino benzoyl glycine in ipsalazine and 4-aminobenzoyl-β-alanine in balsalazide, or azo bond
conjugation of sul-fasalazine with itself to form olsalazine. In addition, the
drug has been covalently conjugated to a polymeric backbone of
polysulfonamidoethylene by azo bond (Figure 15.5).
Polymers
that degrade specifically in the colon have been used for drug targeting by
surface coating to form a barrier to drug release or as matrix systems
embedding the drug substance. For example, azo-linked acrylate copolymers and
poly(ester-ether) copolymers have been used for the delivery of protein and
peptide drugs, and small molecular weight compounds such as ibuprofen, sulfasalazine,
and betamethasone. For embedding the drug in polymer matrices, natural
polysaccharides have been used in oral solid dosage forms to protect the drug
during GI transit and release in the colon on polymer degradation by the
microflora. They offer advantages such as the presence of derivatizable
functional groups and a range of molecular size, in addition to their low
toxicity. The hydrogel (hydrophilic and swelling) prop-erties of these
polymers, however, can lead to the dosage form swelling and disintegration in
the presence of water before reaching the colon. Therefore, these dosage forms
require protection from the aqueous environment during upper GI transit. This
is usually accomplished by the use of protective surface coating or chemical
cross-linking with linkers that are degraded in the colon. Polymers that are
stable in the upper GI tract and degraded by colonic micro-flora include azo
cross-linked synthetic polymers and plant polysaccharides, such as amylose,
pectin, inulin, and guar gum.
A
disadvantage of polymeric coating or embedding approaches for colonic drug
delivery is their dependence on the bacterial microflora in the large
intestine. Although the microflora is fairly constant in the healthy
population, it can be affected by the dietary fermentation precursors, type of
diet consumed, and coadministration of antibiotics. In addition, the nat-ural
polymers are often not available in pure form, which can lead to
physi-cochemical incompatibility with the drug substance and/or inconsistency of
product performance.
pH-dependent dosage forms
pH-sensitive
polymers have been widely used for enteric coating of dosage forms to
facilitate pH-dependent drug release. As the pH increases progres-sively from
stomach (pH 1–2) to small intestine (pH 6–7), and the distal ileum (pH 7–8),
dosage forms can be coated with polymers that dissolve only the aforementioned
specific pH ranges. For colon targeting, the poly-meric coating should be able
to withstand the acidic pH of the stomach and higher pH of the proximal small
intestine, but dissolve in the neutral to slightly basic pH of the terminal
ileum. However, most of the commonly used enteric coating polymeric systems
have a pH threshold of 6.0 or lower for dissolution. These include the
methacrylic acid/methyl methacrylate copolymers, (Eudragits® L100, L-30D,
L100-55), polyvinylacetate phthal-ate (PVAP), hydroxypropyl methylcellulose
phthalate (HPMCP), cellulose acetate phthalate (CAP), and cellulose acetate
trimelliate (CAT). Only Eudragit® S100 and FS 30D have a higher pH threshold of
6.8 and 7.0, respectively.
Eudragit®
S100 coating is used, for example, in the mesalamine (Asacol®, Procter
&Gamble)-delayed release tablets for topical anti-inflammatory action in
the colon. Eudragit® L100 and S100 are copolymers of meth-acrylic acid and
methyl methacrylate with the ratio of carboxyl to ester groups of 1:1 or 1:2,
respectively. The carboxylate groups form salts, lead-ing to polymer
dissolution at basic pH. Drug release from these acrylate polymers also depend on
the plasticizer, nature of the salt in the dissolu-tion medium, and
permeability of the film. Colon-targeted dosage forms utilizing methacrylate
resins for coating or matrix formation have been reported in several molecules
such as bisacodyl, indomethacin, 5-FU, and budesonide.
The
use of pH trigger for drug delivery to the colon, however, has the disadvantage
of inconsistency in dissolution of the polymer at the desired site due to
inter- and intraindividual pH variation, among other factors. For example,
Ashford et al. observed significant variability in the disintegration time and
location of Eudragit® S coated tablets in human volunteers. In addition, based
on GI motility, polymer dissolution can complete toward the end of the ileum or
deep in the colon. In addition, factors such as the presence of short-chain
fatty acids and residues of bile acids in the luminal contents, and the locally
formed fermentation products can reduce the local pH, thus influencing the drug
release mechanism.
Time-dependent drug release
Human
small intestinal transit time for pharmaceutical dosage forms was measured
using gamma scintigraphy and found to be about 3–4 h. Although the transit time
does vary with the amount of food and the type of dosage form, it is less
variable than the gastric emptying time. Timed release of dosage forms to
target the colon are, thus, typically formulated to prevent drug release in the
acidic gastric environment and to prevent the release of drug until 3–4 s after
leaving the acidic gastric environment.
An
example of such timed-release dosage form is the Pulsincap® device. In this
device, the drug formulation is sealed in an impermeable capsule body with a
hydrogel polymer plug. The hard gelatin capsule body may be made insoluble by
exposure to formaldehyde vapor, which cross-links gelatin. The plug expands in
the aqueous GI tract fluid and exits the body, thus releasing drug, after a
time delay determined by the rate of expansion and the length of the plug.
Another
approach utilized a three-layer coated dosage form with an inner coating of an
acid-soluble polymer, Eudragit® E; followed by a water-soluble coat, and the
outer enteric coating of Eudragit® L. An organic acid (succinic acid) was used
as a part of the formulation. On oral administra-tion, the dosage form is
protected in the acidic gastric environment by the enteric coating. In
intestinal conditions, water ingress into the formulation lowers the pH inside
the dosage form by the dissolution of the organic acid.
This,
in turn, causes the inner, acid-labile coat to dissolve, thus releasing the
drug. Drug release rate and lag time is controlled by the coating thickness of
the acid-soluble layer and the amount of organic acid in the formulation. Using
this approach, Fukui et al. prepared timed-release press-coated tablets with
the core tablets containing diltiazem hydrochloride (DIL) and the outer, water
soluble, layer containing phenylpropanolamine hydrochlo-ride (PPA), as a marker
for gastric emptying time. On administration to beagle dogs, the gastric
emptying time and lag time after gastric empty-ing were evaluated by
determining the times at which PPA and DIL first appeared in the plasma, which
were about 4 and 7 h, respectively. The 3 h lag time between the time of
appearance of these drugs in the plasma correlated well with the expected
intestinal transit time.
An
inherent limitation of the time-dependent drug release systems inter-and
intraindividual variability in gastric emptying, and small intestinal and
colonic transit time. This can result in variations in the site of drug release
in the small intestine or within the colon, which can impact drug absorp-tion
as absorption by the transcellular route diminishes in the distal colon.
Osmotically controlled drug delivery systems
Osmotic
DDSs, such as the OROS-CT® system of Alza Corporation, are based on the
incorporation of an osmotic agent, such as a salt, in the dos-age form. The
dosage form is encapsulated in a semipermeable membrane with an orifice for
drug release. On ingestion, osmotic pressure gradient forces the ingress of
water, which leads to the formation of flowable gel in the drug compartment and
generates pressure to force the drug gel out of the orifice at a controlled
rate. Amount of the osmotic agent, rate of water permeation, and size of the
laser-drilled orifice primarily determine the drug release rate. The release
rate can be extended for 4–24 h in the colon and the each osmotic unit is
designed for a 3–4 hpostgastric delay for drug release.
A
modification of the osmotic pump suitable for colonic drug delivery involves
microbially triggered release mechanism. Liu et al. exploited the gelation of
chitosan under acidic conditions and its degradation in the colon to use it as
an osmotic agent and as a pore-forming agent in the impermeable cellulose
acetate membrane. The authors designed a
dosage form containing citric acid and chitosan in the drug containing core,
which had a coating of cellulose acetate and chitosan, followed by an enteric
coat of methacrylic acid/ methyl methacrylate copolymer, Eudragit® L100. As
shown in Figure 15.6a, on reaching the small
intestine, the enteric coat dissolves followed by water permeation into the
core, leading to the formation of a flowable gel through dissolution of citric
acid and swelling of chitosan. However, chitosan in the cellulose acetate
membrane is completely dissolved only in the colonic microenvironment, thus
preventing significant drug release until the dos-age form reaches the colon. Figure 15.6b shows drug (budesonide, used as a model
drug)-release inhibition at gastric and intestinal pH and controlled
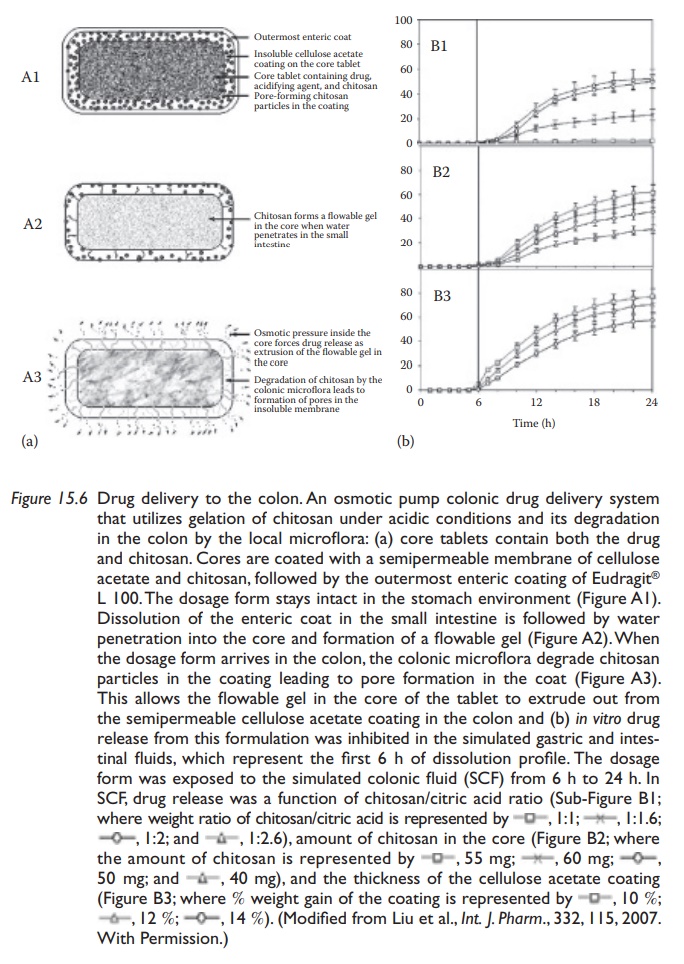
Figure 15.6 Drug delivery to the colon. An osmotic pump colonic drug delivery system that utilizes gelation of chitosan under acidic conditions and its degradation in the colon by the local microflora: (a) core tablets contain both the drug and chitosan. Cores are coated with a semipermeable membrane of cellulose acetate and chitosan, followed by the outermost enteric coating of Eudragit® L 100. The dosage form stays intact in the stomach environment (Figure A1). Dissolution of the enteric coat in the small intestine is followed by water penetration into the core and formation of a flowable gel (Figure A2). When the dosage form arrives in the colon, the colonic microflora degrade chitosan particles in the coating leading to pore formation in the coat (Figure A3). This allows the flowable gel in the core of the tablet to extrude out from the semipermeable cellulose acetate coating in the colon and (b) in vitro drug release from this formulation was inhibited in the simulated gastric and intes-tinal fluids, which represent the first 6 h of dissolution profile.
Related Topics
