Relationship among disintegration, dissolution, and absorption
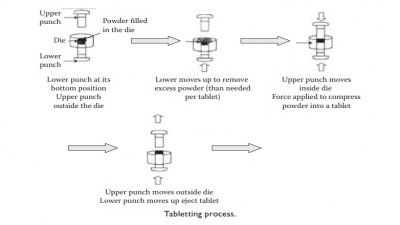
| Home | | Pharmaceutical Drugs and Dosage | | Pharmaceutical Industrial Management |Chapter: Pharmaceutical Drugs and Dosage: Tablets
Oral absorption from the GI tract takes place from the drug in aqueous solution at the site of absorption.
Relationship among
disintegration, dissolution, and absorption
Oral
absorption from the GI tract takes place from the drug in aqueous solution at
the site of absorption. Therefore, a tablet must disintegrate into primary drug
particles and release the drug, that is, the drug should dis-solve in the
fluids at the site of absorption, before absorption can take place (Figure 20.5). However, tablets that are intended for
chewing or SR do not have to undergo disintegration. Excipients for tablet
formulation affect the rates of disintegration, dissolution, and absorption.
Systemic absorption of most products consists of a succession of rate
processes, such as
·
Disintegration of the DP into granules and primary drug
particles
·
Dissolution of the drug from the granules and primary drug
particles in an aqueous environment
·
Absorption of drug solution across cell membranes into the
systemic circulation
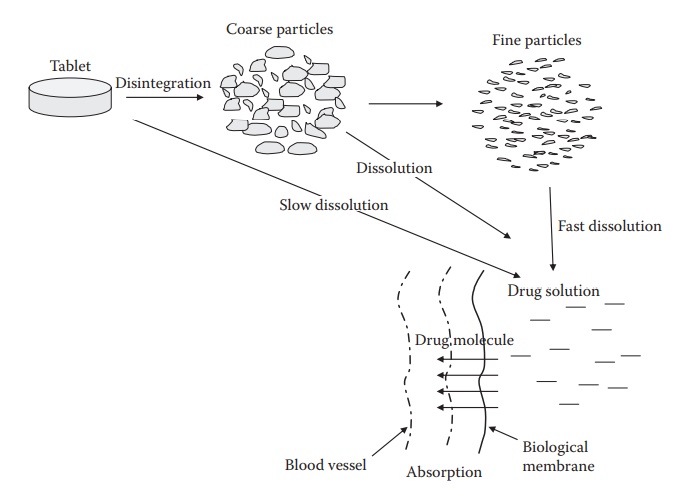
Figure 20.5 Relationship among disintegration, dissolution, and drug absorption
from an intact tablet.
Tablet
disintegration, dissolution, and drug absorption are influenced by
physicochemical properties (e.g., solubility, compactibility, density, and
flow) and stability (e.g., to heat, moisture, and light) of the DS; its
com-patibility with the excipients in the dosage form; site and extent of drug
absorption in the GI tract; and dose. In these processes, the rate at which
drug reaches the circulatory system is determined by the slowest step in the
sequence. Disintegration of a tablet is usually more rapid than drug
dis-solution and absorption. For the drug that has poor aqueous solubility, the
rate at which the drug dissolves (dissolution) is often the slowest step, and
therefore exerts a rate-limiting effect on drug bioavailability. In contrast,
for the drug that has a high aqueous solubility, the dissolution rate is rapid
and the rate at which the drug crosses or permeates cell membranes is the
slowest or rate-limiting step.
Aqueous
solubility and permeability across the intestinal mucosa form the basis of
identifying the relative difficulty in formulating drugs for oral delivery
through what is known as the biopharmaceutics classification sys-tem (BCS). BCS
classifies both the rate of drug permeation and drug solu-bility relative to
the highest dose in two categories: high and low. Thus, all drugs can be
divided into four distinct categories: I—high solubility, high permeability;
II—low solubility, high permeability; III—high solubility, low permeability;
and IV—low solubility, low permeability. This classifi-cation system has been
used to understand and influence (by formulation design) the oral
pharmacokinetics of drugs.