Restriction Fragment Length Polymorphism
| Home | | Biochemistry |Chapter: Biochemistry : Biotechnology and Human Disease
It has been estimated that the genomes of any two unrelated people are 99.5% identical. With 6 billion bp in the diploid human genome, that represents variation in about 30 million bp.
RESTRICTION FRAGMENT LENGTH POLYMORPHISM
It has been estimated
that the genomes of any two unrelated people are 99.5% identical. With 6
billion bp in the diploid human genome, that represents variation in about 30
million bp. These genome variations are the result of mutations that lead to
polymorphisms. A polymorphism is a change in genotype that can result in no
change in phenotype or a change in phenotype that is harmless; causes increased
susceptibility to a disease; or, rarely, causes the disease. It is
traditionally defined as a sequence variation at a given locus (allele) in more
than 1% of a population. Polymorphisms primarily occur in the 98% of the genome
that does not encode proteins (that is, in introns and intergenic regions). A
restriction fragment length polymorphism (RFLP) is a genetic variant that can
be observed by cleaving the DNA into fragments (restriction fragments) with a
restriction enzyme. The length of the restriction fragments is altered if the
variant alters the DNA so as to create or abolish a site of restriction
endonuclease cleavage (a restriction site). RFLP can be used to detect human
genetic variations, for example, in prospective parents or in fetal tissue.
A. DNA variations resulting in restriction fragment length polymorphism
Two types of DNA
variation commonly result in RFLP: single-base changes in the DNA sequence and
tandem repeats of DNA sequences.
1. Single-base changes in DNA: About 90% of human genome
variation comes in the form of single nucleotide polymorphisms (SNPs,
pronounced “snips”), that is, variations that involve just one base (Figure
33.13). The substitution of one nucleotide at a restriction site can render the
site unrecognizable by a particular restriction endonuclease. A new restriction
site can also be created by the same mechanism. In either case, cleavage with
an endonuclease results in fragments of lengths differing from the normal that
can be detected by DNA hybridization (see Figure 33.12). The altered
restriction site can be either at the site of a disease-causing mutation (rare)
or at a site some distance from the mutation. [Note: The HapMap, developed by
The International Haplotype Map Project, is a catalog of common SNPs in the
human genome. The data are being used in genome-wide association studies (GWAS)
to identify those alleles that affect health and disease.]
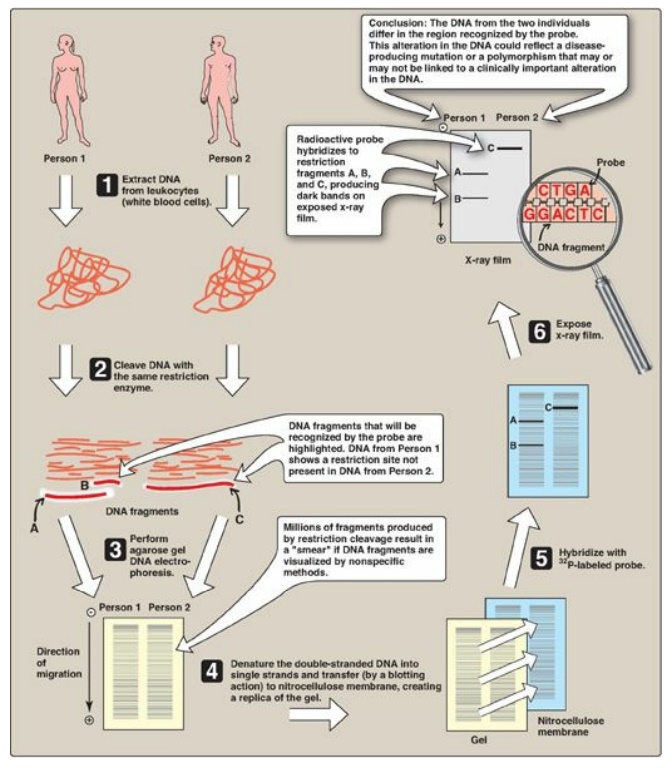
Figure 33.12 Southern blotting
procedure. [Note: Nonradiolabeled probes are now commonly used.]
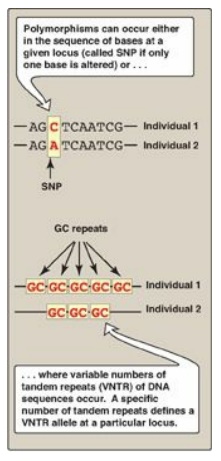
Figure 33.13 Common forms of genetic polymorphism. SNP = single-nucleotide polymorphism. A = adenine; C = cytosine; G = guanine; T = thymine.
2. Tandem repeats: Polymorphism in chromosomal DNA can also arise from the presence of a variable number of tandem repeats [VNTR] see Figure 33.14). These are short sequences of DNA at scattered locations in the genome, repeated in tandem (one after another). The number of these repeat units varies from person to person but is unique for any given individual and, therefore, serves as a molecular fingerprint. Cleavage by restriction enzymes yields fragments that vary in length depending on how many repeated segments are contained in the fragment (Figure 33.14). Many different VNTR loci have been identified and are extremely useful for DNA fingerprint analysis, such as in forensic and paternity cases. It is important to emphasize that these polymorphisms, whether SNP or VNTR, are simply markers, which, in most cases, have no known effect on the structure, function, or rate of production of any particular protein.
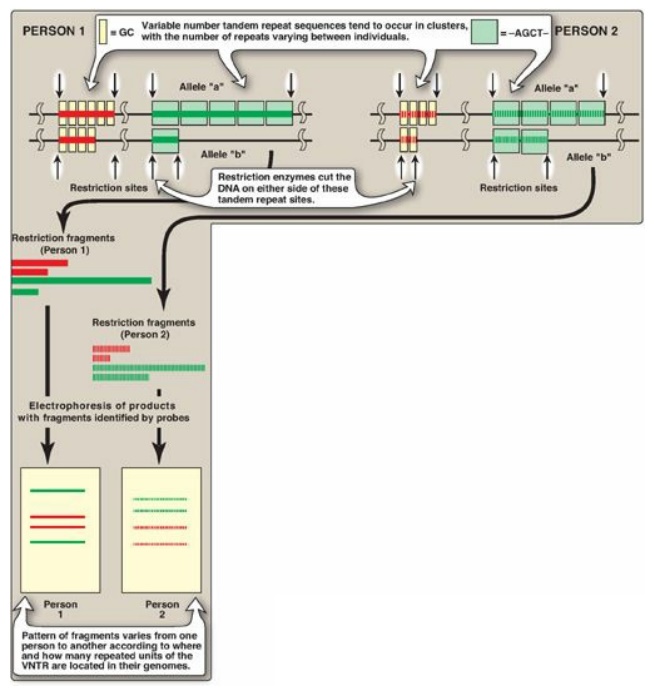
Figure 33.14 Restriction fragment length polymorphism of variable number tandem repeats (VNTR). For each person, a pair of homologous chromosomes is shown.
B. Tracing chromosomes from parent to offspring
If the DNA of an individual has gained a restriction site by base substitution, then enzymic cleavage yields at least one additional fragment. Conversely, if a mutation results in loss of a restriction site, fewer fragments are produced by enzymic cleavage. An individual who is heterozygous for a polymorphism has a sequence variation in the DNA of one chromosome and not in the homologous chromosome. In such individuals, each chromosome can be traced from parent to offspring by determining the presence or absence of the polymorphism.
C. Prenatal diagnosis
Families with a history
of severe genetic disease, such as an affected previous child or near relative,
may wish to determine the presence of the disorder in a developing fetus.
Prenatal diagnosis, in association with genetic counseling, allows for an
informed reproductive decision if the fetus is affected.
1. Methods available: The available diagnostic methods
vary in sensitivity and specificity. Visualization of the fetus, for example,
by ultrasound or fiberoptic devices (fetoscopy), is useful only if the genetic
abnormality results in gross anatomic defects (for example, neural tube
defects). The chemical composition of the amniotic fluid can also provide
diagnostic clues. For example, the presence of high levels of α-fetoprotein is
associated with neural tube defects. Fetal cells obtained from amniotic fluid
or from biopsy of the chorionic villi can be used for karyotyping, which
assesses the morphology of metaphase chromosomes. Staining and cell sorting
techniques permit the rapid identification of trisomies and translocations that
produce an extra chromosome or chromosomes of abnormal lengths. However,
molecular analysis of fetal DNA provides the most detailed genetic picture.
2. Sources of DNA: DNA may be obtained from white
blood cells, amniotic fluid, or chorionic villi (Figure 33.15). For amniotic
fluid, it used to be necessary to grow cells in culture for two to three weeks
in order to have sufficient DNA for analysis. The ability to amplify DNA by PCR
has dramatically shortened the time needed for a DNA analysis.
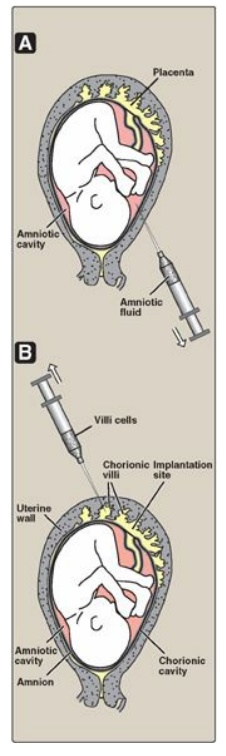
Figure 33.15 Sampling of fetal cells. A. Amniotic fluid. B. Chorionic villus.
3. Direct diagnosis of sickle cell anemia using
RFLP: The
genetic disorders of hemoglobin (Hb) are the most common genetic diseases in
humans. In the case of sickle cell anemia (Figure 33.16), the mutation that
gives rise to the disease is actually one and the same mutation that gives rise
to the polymorphism. Direct detection by RFLP of diseases that result from
point mutations is, however, limited to only a few genetic diseases.
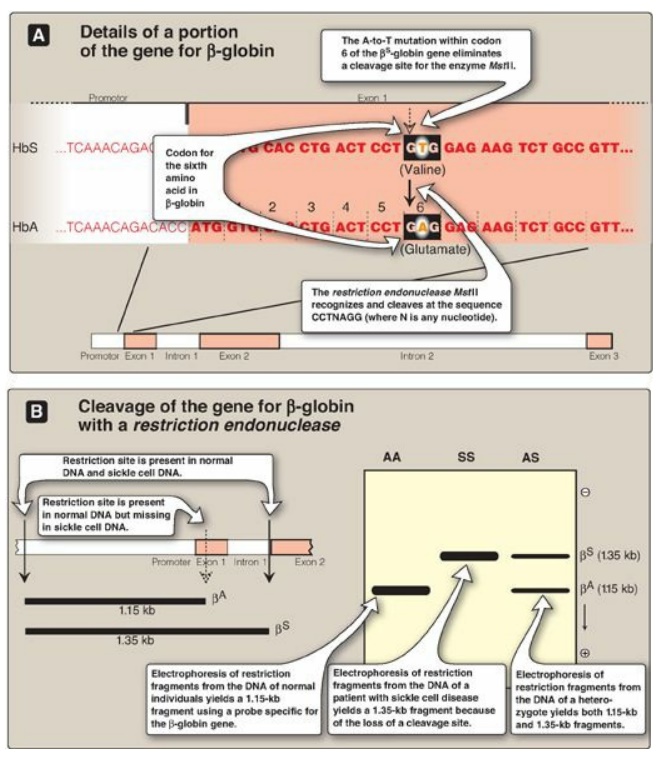
Figure 33.16 Detection of βS-globin mutation. kb = kilobase (1 kb = 1000 base pairs in double-stranded DNA); Hb = hemoglobin.
a. Early efforts to diagnose sickle cell anemia: In the past, prenatal diagnosis of
hemoglobinopathies involved the determination of the amount and kinds of Hb
synthesized in red cells obtained from fetal blood. However, the invasive
procedures to obtain fetal blood have a high mortality rate (approximately 5%),
and diagnosis cannot be carried out until late in the second trimester of
pregnancy when HbS begins to be produced.
b. RFLP analysis: Sickle cell anemia is an example of a genetic disease caused by a point mutation. The sequence altered by the mutation abolishes the recognition site of the restriction endonuclease MstII: CCTNAGG (where N is any nucleotide; see Figure 33.16). Thus, the A-to-T mutation in codon 6 of the bS-globin gene eliminates a cleavage site for the enzyme. Normal DNA digested with MstII yields a 1.15-kb fragment, whereas a 1.35-kb fragment is generated from the bS gene as a result of the loss of one MstII cleavage site. Diagnostic techniques that allow analysis of fetal DNA from amniotic cells or chorionic villus sampling rather than fetal blood have proved valuable because they provide safe, early detection of sickle cell anemia as well as other genetic diseases. [Note: Genetic disorders caused by insertions or deletions between two restriction sites, rather than by the creation or loss of cleavage sites, will also display RFLP.]
4. Indirect, prenatal diagnosis of phenylketonuria
using RFLP: The
gene for phenylalanine hydroxylase (PAH), deficient in phenylketonuria ([PKU]),
is located on chromosome 12. It spans about 90 kb of genomic DNA and contains
13 exons separated by introns (Figure 33.17; see respected section for a
description of exons and introns). Mutations in PAH usually do not directly
affect any restriction endonuclease recognition site. To establish a diagnostic
protocol for this disease, DNA of family members of the affected individual
must be analyzed. The goal is to identify genetic markers (RFLPs) that are
tightly linked to the disease trait. Once these markers are identified, RFLP
analysis can be used to carry out prenatal diagnosis.
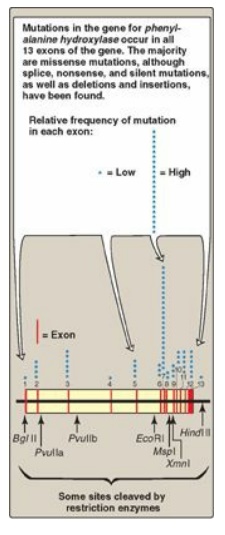
Figure 33.17 The gene for phenylalanine hydroxylase showing 13 exons, restriction sites, and some of the mutations resulting in phenylketonuria.
a. Identification of the gene: Determinining the presence of the mutant gene by identifying the polymorphism marker can be done if two conditions are satisfied. First, if the polymorphism is closely linked to a disease-producing mutation, the defective gene can be traced by detection of the RFLP. For example, if DNA from a family carrying a disease-causing gene is examined by restriction enzyme cleavage and Southern blotting, it is sometimes possible to find an RFLP that is consistently associated with that gene (that is, they show close linkage and are coinherited). It is then possible to trace the inheritance of the gene within a family without knowledge of the nature of the genetic defect or its precise location in the genome. [Note: The polymorphism may be known from the study of other families with the disorder or may be discovered to be unique in the family under investigation.] Second, for autosomal recessive disorders, such as PKU, the presence of an affected individual in the family would aid in the diagnosis. This individual would have the mutation present on both chromosomes, allowing identification of the RFLP associated with the genetic disorder.
b. RFLP analysis: The presence of abnormal genes for
PAH can be shown using DNA polymorphisms as markers to distinguish between
normal and mutant genes. For example, Figure 33.18 shows a typical pattern
obtained when DNA from the white blood cells of a family is cleaved with an
appropriate restriction enzyme and subjected to electrophoresis. The vertical
arrows represent the cleavage sites for the restriction enzyme used. The
presence of a polymorphic site creates fragment “b” in the autoradiogram (after
hybridization with a labeled PAH-cDNA probe), whereas the absence of this site
yields only fragment “a.” Note that Subject II-2 demonstrates that the
polymorphism, as shown by the presence of fragment “b,” is associated with the
mutant gene. Therefore, in this particular family, the appearance of fragment
“b” corresponds to the presence of a polymorphic site that marks the abnormal
gene for PAH. The absence of fragment “b” corresponds to having only the normal
gene. In Figure 33.18, examination of fetal DNA shows that the fetus inherited
two abnormal genes from its parents and, therefore, has PKU.
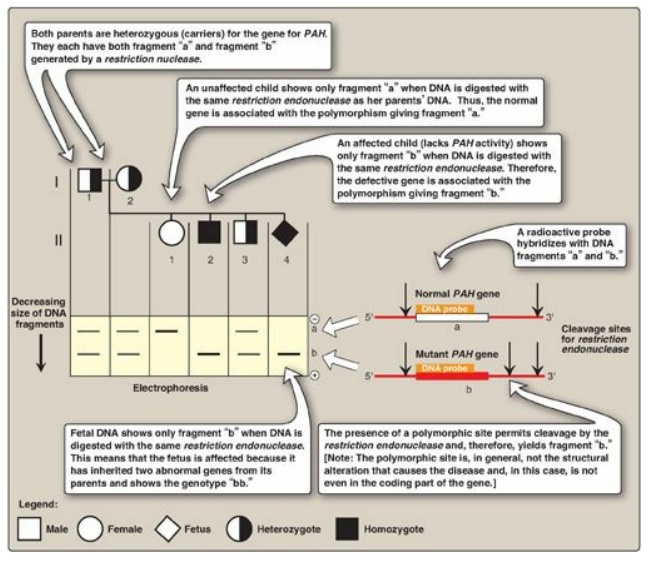
Figure 33.18 Analysis of
restriction fragment length polymorphism in a family with a child affected by
phenylketonuria (PKU), an autosomal recessive disease. The molecular defect in
the gene for phenylalanine hydroxylase (PAH) in the family is not known. The
family wanted to know if the current pregnancy would be affected by PKU.
c. Value of DNA testing: DNA-based testing is useful not only in determining if an unborn fetus is affected by PKU, but also in detecting unaffected carriers of the mutated gene to aid in family planning. [Note: PKU is treatable by dietary restriction of phenylalanine. Early diagnosis and treatment are essential in preventing severe neurologic damage in affected individuals.].
Related Topics
