Biological Factors of Drug
| Home | | Biopharmaceutics and Pharmacokinetics |Chapter: Biopharmaceutics and Pharmacokinetics : Biotransformation of Drugs
Biological Factors: Species Differences, Strain Differences/Pharmacogenetics, Sex Differences, Age, Diet, Altered Physiological Factors, Temporal Factors
BIOLOGICAL FACTORS
Species Differences,
Strain Differences/Pharmacogenetics,
Sex Differences,
Age,
Diet,
Altered Physiological Factors,
Temporal Factors
Species Differences
Screening of new therapeutic molecules to ascertain
their activity and toxicity requires study in several laboratory animal
species. Differences in drug response due to species differences are taken into
account while extrapolating the data to man.
Species differences have been observed in both
phase I and phase II reactions. In phase I reactions, both qualitative and
quantitative variations in the enzyme and their activity have been observed. An
example of this is the metabolism of amphetamine and ephedrine. In men and
rabbit, these drugs are predominantly metabolised by oxidative deamination
whereas in rats the aromatic oxidation is the major route. In phase II
reactions, the variations are mainly qualitative and characterized either by
the presence of, or complete lack of certain conjugating enzymes; for example,
in pigs, the phenol is excreted mainly as glucuronide whereas its sulphate
conjugate dominates in cats. Certain birds utilize ornithine for conjugating
aromatic acids instead of glycine.
Strain Differences/Pharmacogenetics
Enzymes influencing metabolic reactions are under
the genetic control. Just as the differences in drug metabolising ability
between different species are attributed to genetics, so also are the
differences observed between strains of the same animal species. A study of inter-subject variability in drug response (due to differences in, for
example, rate of biotransformation) is called as pharmacogenetics. The inter-subject variations in drug biotransformation may either be
monogenically or polygenically controlled. A polygenic control has
been observed in studies in twins. In identical twins (monozygotic), very
little or no difference in the
metabolism of phenylbutazone, dicoumarol and antipyrine was detected but large
variations were apparent in fraternal twins (dizygotic; twins developed from
two different eggs) for the same drugs.
Differences observed in the metabolism of a drug among different races
are called as ethnic variations. Such a variation may be monomorphic or
polymorphic. When a unimodal frequency
distribution is observed in the entire population, the variations are called as
continuous or
monomorphic; for example, the entire human race acetylate PABA and PAS to
only a small extent. A polymodal
distribution is indicative of discontinuous variation (polymorphism).
An example of polymorphism is the acetylation of isoniazid (INH) in humans. A bimodal population
distribution was observed comprising of slow acetylator or inactivator
phenotypes (metabolise INH slowly) and rapid acetylator or inactivator
phenotypes (metabolise INH rapidly) (see
Table 5.6.).
TABLE 5.6
Ethnic Variations in the N-Acetylation of Isoniazid
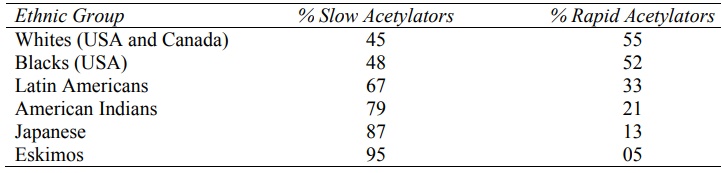
Approximately equal percent of slow and rapid
acetylators are found among whites and blacks whereas the slow acetylators
dominate Japanese and Eskimo populations. Dose adjustments are therefore
necessary in the latter groups since high levels of INH may cause peripheral
neuritis. Other drugs known to exhibit pharmacogenetic differences in
metabolism are debrisoquine, succinyl choline, phenytoin, dapsone and
sulphadimidine.
Sex Differences
Sex related differences in the rate of metabolism
could be attributed to regulation of such processes by sex hormones since
variations between male and female are generally observed following puberty.
Such sex differences are widely studied in rats; the male rats have greater
drug metabolising capacity. In humans, women metabolise benzodiazepines slowly
than men and several studies show that women on contraceptive pills metabolise
a number of drugs at a slow rate.
Age
Differences in the drug metabolic rate in different
age groups are mainly due to variations in the enzyme content, enzyme activity
and haemodynamics.
·
In neonates (upto 2 months), the
microsomal enzyme system is not fully developed and many drugs are
biotransformed slowly; for example, caffeine has a half-life of 4 days in
neonates in comparison to 4 hours in adults. A major portion of this drug is
excreted unchanged in urine by the neonates. Conjugation with sulphate is well
developed (paracetamol is excreted mainly as sulphate) but glucuronidation
occurs to a very small extent. As a result, hyperbilirubinaemia precipitates
kernicterus and chloramphenicol leads to cyanosis or Gray baby syndrome in new
born. Similarly, sulphonamides cause renal toxicity and paracetamol causes
hepatotoxicity.
·
Infants (between 2 months and one
year) show almost a similar profile as neonates in metabolising drugs with
improvement in the capacity as age advances and enzyme activity increases.
·
Children (between one year and 12
years) and older infants metabolise several drugs much more rapidly than adults
as the rate of metabolism reaches a maximum somewhere between 6 months and 12
years of age. As a result, they require large mg/Kg doses in comparison to
adults; for example, the theophylline half-life in children is two-third of
that in adults.
·
In very elderly persons, the
liver size is reduced, the microsomal enzyme activity is decreased and hepatic
blood flow also declines as a result of reduced cardiac output all of which
contribute to decreased metabolism of drugs. Drug conjugation however remains
unaffected.
Diet
The enzyme content and activity is altered by a number
of dietary components. In general –
·
Low protein diet decreases and
high protein diet increases the drug metabolising ability. This is because the
enzyme synthesis is promoted by protein diet which also raises the level of
amino acids for conjugation with drugs.
·
The protein-carbohydrate ratio in
the diet is also important; a high ratio increases the microsomal mixed
function oxidase activity.
·
Fat free diet depresses
cytochrome P-450 levels since phospholipids, which are important components of
microsomes, become deficient.
·
Dietary deficiency of vitamins
(e.g. vitamin A, B2, B3, C and E) and minerals such as
Fe, Ca, Mg, Cu and Zn retard the metabolic activity of enzymes.
·
Grapefruit inhibits metabolism of
many drugs and improve their oral availability.
·
Starvation results in decreased
amount of glucuronides formed than under normal conditions.
·
Malnutrition in women results in
enhanced metabolism of sex hormones.
·
Alcohol ingestion results in a
short-term decrease followed by an increase in the enzyme activity.
Altered Physiological Factors
Pregnancy: Studies in animals have shown
that the maternal drug metabolising ability (of both phase I and phase II reactions) is reduced during the later
stages of pregnancy. This was suggested as due to high levels of steroid
hormones in circulation during pregnancy. In women, the metabolism of promazine
and pethidine is reduced during pregnancy or when receiving oral
contraceptives. Higher rate of hepatic metabolism of anticonvulsants during
pregnancy is thought to be due to induction of drug metabolising enzymes by the
circulating progesterone.
Hormonal Imbalance: The influence of sex hormones on
drug metabolism has already been
discussed. The effect of other hormones is equally complex. Higher levels of
one hormone may inhibit the activity of few enzymes while inducing that of
others. Adrenolectomy, thyroidectomy and alloxan induced diabetes in animals
showed impairment in the enzyme activity with a subsequent fall in the rate of
metabolism. A similar effect was observed with pituitary growth hormone. Stress
related changes in ACTH levels also influence drug biotransformation.
Disease States: As liver is the primary site for
metabolism of most drugs, all pathologic
conditions associated with it result in enhanced half-lives of almost all
drugs. Thus, a reduction in hepatic drug metabolising ability is apparent in
conditions such as hepatic carcinoma, hepatitis, cirrhosis, obstructive
jaundice, etc. Biotransformations such as glycine conjugation of salicylates,
oxidation of vitamin D and hydrolysis of procaine which occur in kidney, are
impaired in renal diseases. Congestive cardiac failure and myocardial
infarction which result in a decrease in the blood flow to the liver, impair
metabolism of drugs having high hepatic extraction ratio e.g. propranolol and
lidocaine. In diabetes, glucuronidation is reduced due to decreased
availability of UDPGA.
Temporal Factors
Circadian Rhythm: Diurnal variations or variations in the enzyme activity with light cycle is called as circadian rhythm in drug metabolism. It has been observed that the enzyme
activity is maximum during early morning (6 to 9 a.m.) and minimum in late
afternoon (2 to 5 p.m.) which was suggested to correspond with the high and low
serum levels of corticosterone (the serum corticosterone level is dependent
upon the light-dark sequence of the day). Clinical variation in therapeutic
effect of a drug at different times of the day is therefore apparent. The study of variations in drug response as
influenced by time is called as chronopharmacology. Time dependent change in drug kinetics is known as chronokinetics. Drugs such as
aminopyrine, hexobarbital and imipramine showed diurnal variations in rats. The half-life of metyrapone was shown to be
2.5 times longer during the night than in the day, in rats.
Related Topics
