Drug Interactions
| Home | | Biopharmaceutics and Pharmacokinetics |Chapter: Biopharmaceutics and Pharmacokinetics : Protein Binding of Drugs
Competition Between Drugs for the Binding Sites (Displacement Interactions)
DRUG INTERACTIONS
Competition Between Drugs for the Binding Sites (Displacement Interactions)
When two or more drugs can bind to the same site, competition between them for interaction with the binding site results. If one of the drugs (drug A) is bound to such a site, then administration of another drug (drug B) having affinity for the same site results in displacement of drug A from its binding site. Such a drug-drug interaction for the common binding site is called as displacement interaction. The drug A here is called as the displaced drug and drug B as the displacer. Warfarin and phenylbutazone have same degree of affinity for HSA. Administration of phenylbutazone to a patient on warfarin therapy results in displacement of latter from its binding site. The free warfarin may cause adverse hemorrhagic reactions which may be lethal. Phenylbutazone is also known to displace sulphonamides from their HSA binding sites. Displacement interactions can result in unexpected rise in free concentration of the displaced drug which may enhance clinical response or toxicity. Even a drug metabolite can affect displacement interaction.
Clinically significant interactions will result when:
I. The displaced drug (e.g. warfarin) —
1. Is more than 95% bound.
2. Has a small volume of distribution (less than 0.15 L/Kg).
3. Shows a rapid onset of therapeutic or adverse effects.
4. Has a narrow therapeutic index.
II. The displacer drug (e.g. phenylbutazone) —
1. Has a high degree of affinity as the drug to be displaced.
2. Competes for the same binding sites.
3. The drug/protein concentration ratio is high (above 0.10).
4. Shows a rapid and large increase in plasma drug concentration.
It will be worthwhile to mention here that, both the concentration of the displacer drug and its affinity for the binding site with respect to that of the drug to be displaced, will determine the extent to which displacement will occur.
For a drug that is 95% bound, a displacement of just 5% of the bound drug results in a 100% rise in free drug concentration. If the displaced drug has a small volume of distribution, it remains confined to the blood compartment and shows serious toxic responses. On the contrary, if such a drug has a large Vd, it redistributes into a large volume of body fluids and clinical effects may be negligible or insignificant. The increase in free drug concentration following displacement also makes it more available for elimination by the liver and the kidneys (Fig. 4.3). If the drug is easily metabolisable or excretable, its displacement results in significant reduction in elimination half-life.
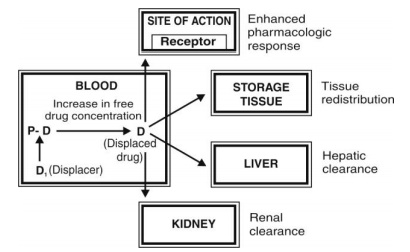
Fig. 4.3. Fate of a drug after displacement interaction
Displacement also becomes insignificant with the use of more selective, potent, low dose drugs.
Besides the direct displacement interaction discussed above, indirect interactions are also possible. For example, the use of heparin as an anticoagulant activates lipoprotein lipase, an enzyme which metabolises triglycerides to free fatty acids. Heparin co-administration with drugs has also been shown to result in decreased protein binding of propranolol, quinidine, etc. via its effects on fatty acid levels.
Competition Between Drugs and Normal Body Constituents
Among the various normal body constituents, the free fatty acids are known to interact with a number of drugs that bind primarily to HSA. The free fatty acid level is increased in several physiologic (fasting), pathologic (diabetes, myocardial infarction, alcohol abstinence) and pharmacologically induced conditions (after heparin and caffeine administration). The fatty acids, which also bind to albumin, influence binding of several benzodiazepines and propranolol (decreased binding) and warfarin (increased binding). Bilirubin binding to HSA can be impaired by certain drugs and is of great concern in neonates whose BBB and bilirubin metabolising capacity are not very efficient. Acidic drugs such as sodium salicylate, sodium benzoate and sulphonamides displace bilirubin from its albumin-binding site. The free bilirubin is not conjugated by the liver of the neonates and thus crosses the BBB and precipitates the condition called as kernicterus (characterized by degeneration of brain and mental retardation).
Allosteric Changes in Protein Molecule
This is yet another mechanism by which drugs can affect protein-binding interactions. The process involves alteration of the protein structure by the drug or its metabolite thereby modifying its binding capacity. The agent that produces such an effect is called as allosteric effector, e.g. aspirin acetylates the lysine fraction of albumin thereby modifying its capacity to bind NSAIDs like phenylbutazone (increased affinity) and flufenamic acid (decreased affinity).
Related Topics
