Significance of Protein/Tissue Binding of Drugs
| Home | | Biopharmaceutics and Pharmacokinetics |Chapter: Biopharmaceutics and Pharmacokinetics : Protein Binding of Drugs
Significance of Protein/Tissue Binding of Drugs : Absorption, Systemic Solubility of Drugs, Distribution, Tissue Binding, Apparent Volume of Distribution and Drug Storage, Elimination, Displacement Interactions and Toxicity, Diagnosis, Therapy and Drug Targeting
SIGNIFICANCE OF PROTEIN/TISSUE BINDING OF DRUGS
Absorption
The absorption equilibrium is attained by transfer
of free drug from the site of administration into the systemic circulation and
when the concentration in these two compartments become equal. Following
equilibrium, the process may stop. However, binding of the absorbed drug to
plasma proteins decreases free drug concentration and disturbs such equilibrium.
Thus, sink conditions and the concentration gradient are re-established which
now act as the driving force for further absorption. This is particularly
useful in case of ionised drugs which are transported with difficulty.
Systemic Solubility of Drugs
Water insoluble drugs, neutral endogenous
macromolecules such as heparin and several steroids and oil soluble vitamins
are circulated and distributed to tissues by binding especially to lipoproteins
which act as a vehicle for such hydrophobic compounds.
Distribution
Plasma protein binding restricts the entry of drugs
that have specific affinity for certain tissues. This prevents accumulation of
a large fraction of drug in such tissues and thus, subsequent toxic reactions.
Plasma protein-drug binding thus favours uniform distribution of drugs
throughout the body by its buffer function (maintains equilibrium between the
free and the bound drug). A protein bound drug in particular does not cross the
BBB, the placental barrier and the glomerulus.
Tissue Binding, Apparent Volume of Distribution and Drug Storage
A drug that is extensively bound to blood
components remains confined to blood. Such a drug has a small volume of
distribution. A drug that shows extravascular tissue binding has a large volume
of distribution. A tissue or blood component that has great affinity for a
particular drug acts as a depot or storage site for that drug; for example,
RBC is a storage site for the lipophilic compound tetrahydrocannabinol.
The relationship between tissue-drug binding and
apparent volume of distribution can be established as follows –
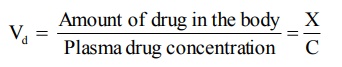
Vd = Amount of drug in the body / Plasma
drug concentration = X/C (4.1)
or, the amount of drug in the body, X = Vd
C (4.2)
Similarly, we can write,
Amount of drug in plasma = Vp C (4.3)
and, Amount of drug in extravascular tissues Vt
Ct (4.4)
The total amount of drug in the body is the sum of
amount of drug in plasma and the amount of drug in extravascular tissues. Thus,
Vd C = Vp C + Vt +
Ct (4.5)
Where, Vd = apparent volume of
distribution of drug
Vp = volume of plasma
Vt = volume of extravascular tissues
Ct = tissue drug concentration
Dividing equation 4.5 with C we get:
Vd = Vp + Vt (4.6)
The fraction of drug unbound (fu) in
plasma is given as:
fu = Concentration of unbound drug in
plasma / Total plasma drug concentration = Cu / C (4.7)
Similarly, fraction of drug unbound to tissues is:
fut = Cut / Ct (4.8)
Assuming that at distribution equilibrium, the
unbound or free drug concentration in plasma equals that in extravascular
tissues i.e. Cu = Cut, equations 4.7 and 4.8 can be
combined to give:
Ct / C = fu / fut (4.9)
Substitution of equation 4.9 in 4.6 yields:
Vd = Vp + [Vt fu]/fut
(4.10)
From equation 4.10 it is clear that greater the
unbound or free concentration of drug in plasma, larger its Vd.
Elimination
Only the unbound or free drug is capable of being
eliminated. This is because the drug-protein complex cannot penetrate into the
metabolising organ (liver). The large molecular size of the complex also
prevents it from getting filtered through the glomerulus. Thus, drugs which are
more than 95% bound are eliminated slowly i.e. they have long elimination
half-lives; for example, tetracycline, which is only 65% bound, has an
elimination half-life of 8.5 hours in comparison to 15.1 hours of doxycycline
which is 93% bound to plasma proteins. However, penicillins have short
elimination half-lives despite being extensively bound to plasma proteins. This
is because rapid equilibration occurs between the free and the bound drug and
the free drug is equally rapidly excreted by active secretion in renal tubules.
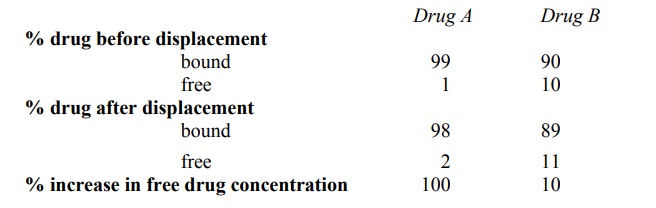
Displacement Interactions and Toxicity
As stated earlier, displacement interactions are
significant in case of drugs which are more than 95% bound. This is explained
from the example given in Table 4.4. A displacement of just 1% of a 99% bound
drug results in doubling of the free drug concentration i.e. a 100% rise. For a
drug that is bound to a lesser extent e.g. 90%, displacement of 1% results in
only a 10% rise in free drug concentration which may be insignificant
clinically.
TABLE 4.4.
Influence of Percent Binding and Displacement on Change in Free
Concentration of Drugs
Kernicterus in infants is an example of a disorder
caused by displacement of bilirubin from albumin binding sites by the NSAIDs
ans sulphonamides. Another example discussed earlier was that of interaction
between warfarin and phenylbutazone. Yet another example of displacement is
that of digoxin with quinidine. Digoxin represents a drug with a large volume
of distribution (i.e. shows extensive extravascular tissue binding). Since
displacement interactions may precipitate toxicity of displaced drug, a
reduction in its dose may be called for. This may become necessary for a drug
having a small Vd such as warfarin since displacement can result in
a large increase in free drug concentration in plasma. With a drug of large Vd
such as digoxin, even a substantial increase in the degree of displacement of drug
in plasma may not effect a large increase in free drug concentration and dose
adjustment may not be required. This is for two reasons—one, only a small
fraction of such a drug is present in plasma whereas most of it is localized in
extravascular tissues, and secondly, following displacement, the free drug,
because of its large Vd, redistributes in a large pool of extravascular
tissues. The extent to which the free plasma drug concentration of drugs with
different Vd values will change when displaced, can be computed from
Equation 4.10.
Diagnosis
The chlorine atom of chloroquine when replaced with
radiolabelled I-131 can be used to visualize melanomas of the eye since
chloroquine has a tendency to interact with the melanin of eyes. The thyroid
gland has great affinity for iodine containing compounds; hence any disorder of
the same can be detected by tagging such a compound with a radioisotope of
iodine.
Therapy and Drug Targeting
The binding of drugs to lipoproteins can be used
for site-specific delivery of hydrophilic moieties. This is particularly useful
in cancer therapies since certain tumour cells have greater affinity for LDL
than normal tissues. Thus, binding of a suitable antineoplastic to it can be
used as a therapeutic tool. HDL is similarly transported more to adrenal and
testes. An example of site-specific drug delivery in cancer treatment is that
of oestramustine. Oestradiol binds selectively and strongly to prostrate and
thus prostrate cancer can be treated by attaching nitrogen mustard to
oestradiol for targeting of prostrate glands. Drug targeting prevents normal
cells from getting destroyed.
Related Topics
