Oxidative Reactions
| Home | | Biopharmaceutics and Pharmacokinetics |Chapter: Biopharmaceutics and Pharmacokinetics : Biotransformation of Drugs
Oxidative reactions are the most important and most common metabolic reactions.
PHASE I REACTIONS
OXIDATIVE REACTIONS
Oxidative reactions are the most important and most
common metabolic reactions. Almost all drugs that undergo phase I
biotransformation undergo oxidation at some stage or the other. A simple reason
for oxidation being a predominant reaction is that energy in animals is
primarily derived by oxidative combustion of organic molecules containing
carbon and hydrogen atoms.
Oxidative reactions increase hydrophilicity of
xenobiotics by introducing polar functional groups such as—OH. Such a polar
metabolite can thus rapidly undergo phase II reaction or is excretable by the
kidneys.
Oxidation of xenobiotics is non-specifically
catalysed by a number of enzymes located in the microsomes. Such enzymes
require both molecular oxygen (O2)
and the reducing agent NADPH to
effect reaction. They are therefore referred to as the mixed-function oxidases. The overall stoichiometry of this
reaction involving the substrate RH which yields the product ROH, is given by
the following equation:
RH + O2 + NADPH + H+ → ROH + H2O
+ NADP+
where NADPH = reduced nicotinamide adenine
dinucleotide phosphate.
Since only one oxygen atom from the molecular
oxygen (dioxygen or O2) is incorporated in the product formed, the
mixed function oxidases are also called as monooxygenases.
Quite often, the product of such a reaction contains a hydroxyl function;
hence, the enzymes are sometimes also called as hydroxylases.
The multienzyme mixed function oxidase system,
located in the endoplasmic reticulum of hepatic cells, is composed of an
electron transfer chain consisting of 3 components:
1. A heme protein known as cytochrome
P-450 (CYP-450), which is actually a family of enzymes. It is a terminal
oxidase. Its function is to transfer an oxygen atom to the substrate RH and
convert it to ROH.
2. A second enzyme, the flavoprotein known as cytochrome P-450 reductase (or cytochrome c reductase) which is
NADPH-dependent. It functions as an electron carrier, catalysing the reduction of cytochrome P-450 to the
ferrous form by transferring an electron from NADPH.
3. A heat
stable lipid component known as phosphatidylcholine.
Its function is to facilitate electron transfer from NADPH to cytochrome P-450.
Magnesium ions are also required for maximal
activity of mixed function oxidases.
The most important component of mixed function
oxidases is the cytochrome P-450 since it binds to the substrate and activates
oxygen. The reduced form of this enzyme (Fe++) binds with carbon
monoxide to form a complex that shows maximum absorption at 450 nm, hence the
name. The mechanism of cytochrome P-450 catalysed metabolism of xenobiotics is
depicted in the redox cycle in Fig. 5.3.
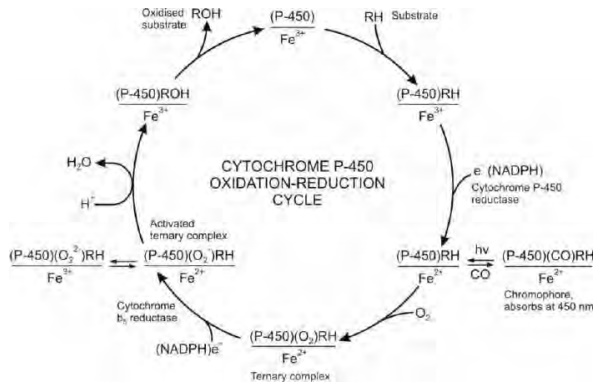
Fig. 5.3. Cytochrome P-450
oxidation-reduction cycle
The various steps
in the oxidation of xenobiotics are:
1. Binding of the substrate (RH)
to the oxidised form of the cytochrome P-450 (Fe+++) to form a
complex.
2. A one-electron transfer from
NADPH to the complex by cytochrome P-450 reductase to form reduced (Fe++)
P-450—substrate complex. This step is considered as the rate-limiting step in
the overall oxidation of xenobiotics.
3. The reduced enzyme-substrate
complex combines with a molecule of oxygen to form a ternary complex.
4. The ternary complex combines
with a second electron supplied by NADH in presence of enzyme cytochrome b5
reductase to form a ternary activated oxygen—P-450—
substrate complex.
5. One atom of oxygen from the
activated oxygen complex is transferred to the substrate to yield the oxidised
product and the other atom forms water. The free oxidised form of cytochrome
P-450 is now ready to attach to yet another molecule of substrate.
Oxidation of Aromatic Carbon Atoms (Aromatic Hydroxylation)
This reaction proceeds via formation of a reactive intermediate arene oxide (epoxide),
which in most cases undergoes rearrangement to yield arenols, and in some cases
catechols and glutathione conjugates.
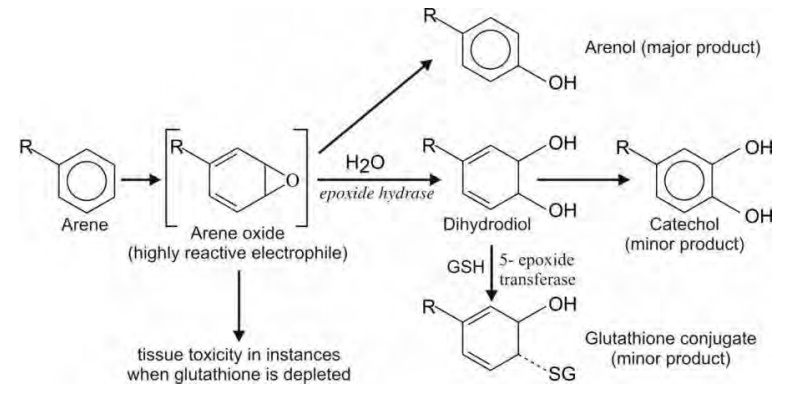
The arene oxide intermediate is highly reactive and
known to be carcinogenic or cytotoxic in some instances, e.g. epoxides of
bromobenzene and benzo(a)pyrene.
Monosubstituted benzene derivatives can be
hydroxylated at ortho-, meta- or para-positions but para-hydroxylated product
is most common, e.g. conversion of acetanilide to paracetamol, and
phenylbutazone to oxyphenbutazone.
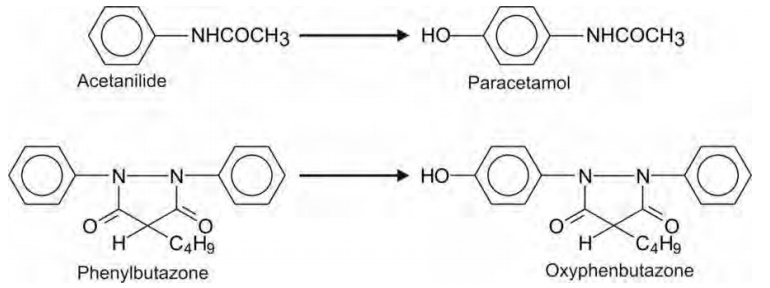
Such a reaction is favoured if the substituent is
an activating group (electron rich) like the amino group. Deactivating or
electron withdrawing groups such as carboxyl and sulphonamide retard or prevent
aromatic hydroxylation, e.g. probenecid.
Oxidation of Olefins
Oxidation of nonaromatic carbon-carbon double bonds
is analogous to aromatic hydroxylation i.e. it proceeds via formation of epoxides to yield 1,2-dihydrodiols. A better known
example of olefinic oxidation is conversion of carbamazepine to
carbamazepine-10,11-epoxide; the latter is converted to corresponding
trans-10,11-dihydrodiol.
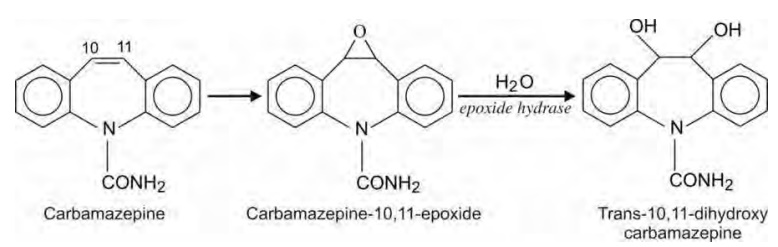
Olefinic hydroxylation differs from aromatic
hydroxylation in that their epoxides are stable and detectable which also
indicate that they are not as reactive as aromatic epoxides. However, an
important example where the olefin epoxide is highly reactive is that of
aflatoxin B1. It is known as the most potent carcinogen (causes
hepatic cancer).
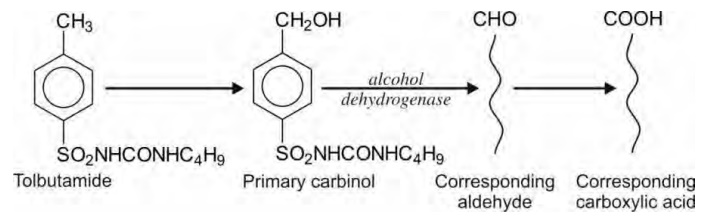
Oxidation of Benzylic Carbon Atoms
Carbon atoms attached directly to the aromatic
rings (benzylic carbon atoms) are hydroxylated to corresponding carbinols. If
the product is a primary carbinol, it is further oxidised to aldehydes and then
to carboxylic acids, e.g. tolbutamide. A secondary carbinol is converted to
ketone.
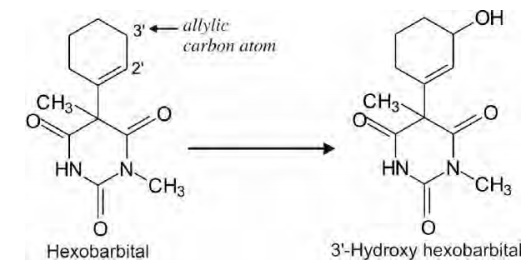
Oxidation of Allylic Carbon Atoms
Carbon atoms adjacent to olefinic double bonds (are
allylic carbon atoms) also undergo hydroxylation in a manner similar to
benzylic carbons, e.g. hydroxylation of hexobarbital to 3'-hydroxy hexobarbital.
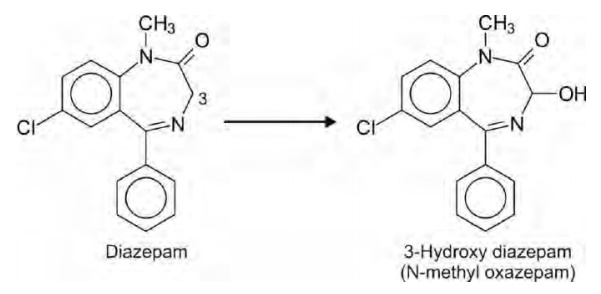
Oxidation of Carbon Atoms Alpha to Carbonyls and Imines
Several benzodiazepines contain a carbon atom (C-3)
alpha to both carbonyl (C=O) and imino (C=N) functions which readily undergoes
hydroxylation, e.g. diazepam.
Oxidation of Aliphatic Carbon Atoms (Aliphatic Hydroxylation)
Alkyl or aliphatic carbon atoms can be hydroxylated
at two positions - at the terminal methyl group (called as -oxidation) and the penultimate carbon atom (called as -1 oxidation) of which the latter
accounts for the major product, e.g. valproic acid. Hydroxylation at other
carbon atoms in long chain compounds is less common.
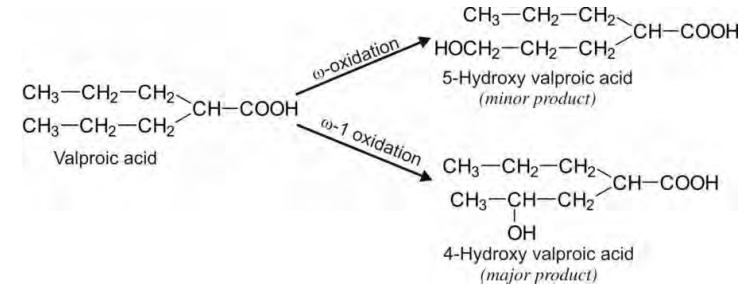
Terminal hydroxylation of methyl group yields
primary alcohol which undergoes further oxidation to aldehyde and then to
carboxylic acid quite rapidly. Penultimate carbon atom can be secondary or
tertiary of which the latter type is also equally reactive, e.g. ibuprofen.
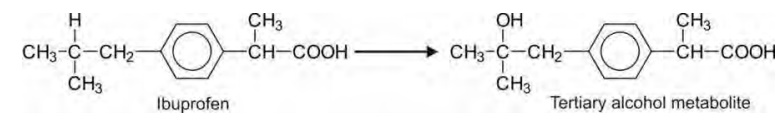
The ω-1
oxidations of secondary and tertiary penultimate carbons yield corresponding
alcohols. The secondary alcohol products seldom undergo further oxidation to
ketones as the latter are less hydrophilic. Further oxidation of tertiary
alcohol products is improbable.
Hydroxylation of aliphatic side chains attached to
an aromatic ring generally occurs at benzylic methylene groups (1') which can
be considered as ω -1 oxidation, e.g. parbendazole.
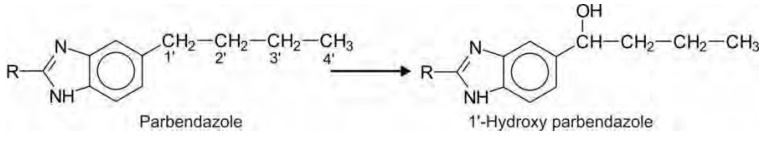
Oxidation of Alicyclic Carbon Atoms (Alicyclic Hydroxylation)
Cyclohexane (alicyclic) and piperidine (nonaromatic
heterocycle) rings are commonly found in a number of molecules, e.g.
acetohexamide and minoxidil respectively. Such rings are generally hydroxylated
at C-3 or C-4 positions.

Oxidation of Carbon-Heteroatom Systems
Biotransformation of C-N, C-O and C-S systems
proceeds in one of the two ways –
1. Hydroxylation of carbon atom attached to the
heteroatom and subsequent cleavage at
carbon-heteroatom bond, e.g. N-, O- and S- dealkylation, oxidative deamination
and desulphuration.
2. Oxidation of the heteroatom itself, e.g.
N- and S-oxidation.
Oxidation of Carbon-Nitrogen Systems
1. N-Dealkylation: Alkyl groups attached directly to
nitrogen atom in nitrogen bearing compounds
are capable of undergoing N-dealkylation reactions, e.g. secondary and tertiary
aliphatic and aromatic amines, tertiary alicyclic amines and N-substituted
amides and hydrazines. Since N-dealkylation of amines yield amines and amides
yield amides, the reaction is said to undergo without any change in the state
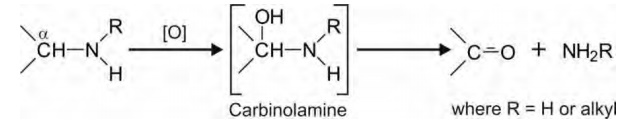
of oxidation. It is however the removed alkyl group that is oxidised. Mechanism
of N-dealkylation involves oxidation of α- carbon
to generate an intermediate carbinolamine
which rearranges by cleavage of C-N bond to yield the N-dealkylated product and
the corresponding carbonyl of the alkyl group (a primary alkyl is transformed
to aldehyde and a secondary alkyl to ketone).
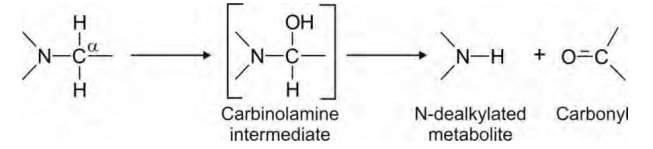
Tertiary nitrogen is more rapidly dealkylated in
comparison to secondary nitrogen because of its higher lipid solubility. Thus,
one alkyl from a tertiary nitrogen compound is removed rapidly and the second
one slowly. Small alkyl groups like methyl, ethyl, n-propyl, isopropyl, etc.
are removed rapidly. N-dealkylation of t-butyl group is not possible because
the -carbon cannot be hydroxylated. Such groups are, however, removed via initial hydroxylation of one of the
methyl groups to alcohol.
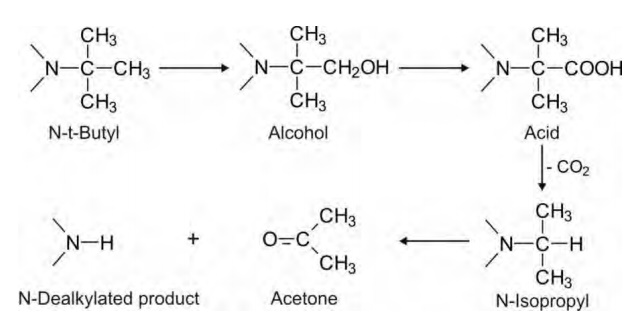
Tertiary nitrogen attached to different alkyl
groups undergoes dealkylation by removal of smaller alkyl group first. A
representative example of each of the chemical classes of compounds capable of
undergoing N-dealkylation is given below.
Secondary aliphatic amines e.g.
methamphetamine.
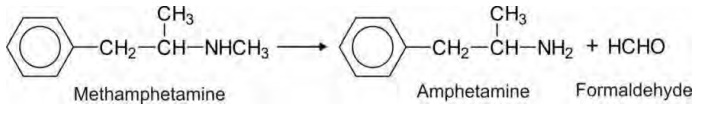
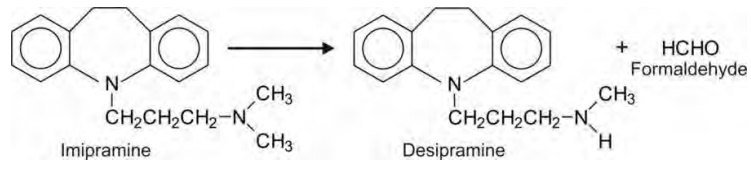
Tertiary aliphatic amines e.g.
imipramine.
Secondary and tertiary aromatic amines are rare
among therapeutic agents.
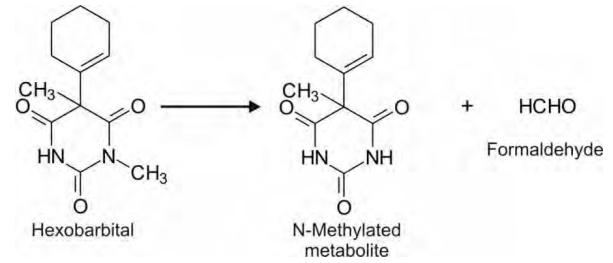
Tertiary alicyclic amines (nonaromatic heterocycle) e.g. hexobarbital.
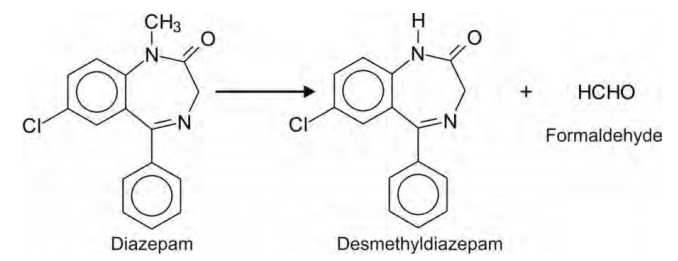
Amides e.g. diazepam.
Hydrazines e.g. iproniazid.
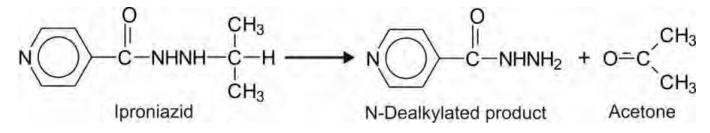
2. Oxidative Deamination: Like
N-dealkylation, this reaction also proceeds
via the carbinolamine
pathway but here the C-N bond cleavage occurs at the bond that links amino
group to the larger portion of the drug molecule.
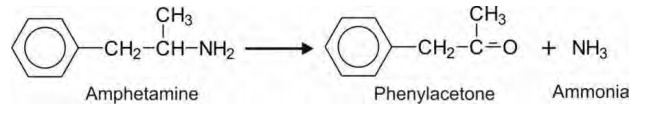
Thus, oxidative deamination is converse of N-dealkylation
in terms of product formed — the carbonyl product retains a large portion of
the parent structure and the amines formed are simple, e.g. NH3.
Actually speaking, both the reactions are same, the criterion for
differentiation being the relative size of the products.
Primary aliphatic amines readily undergo
deamination, e.g. amphetamine, while secondary and tertiary amines are
deaminated only when bulky groups are attached to nitrogen, e.g. propranolol.
Primary amine metabolites formed by N-dealkylation
or decarboxylation also undergo deamination. Deamination is inhibited by the
absence of -hydrogen, e.g. phentermine. Aromatic and alicyclic amines are
resistant to deamination.
3. N-Oxide Formation: N-oxides
are formed only by the nitrogen atoms having basic properties. Thus, amines can form N-oxides but amides cannot.
Generally, the tertiary amines yield N-oxides. Four categories of tertiary
amines that form N-oxides are—
(i) Aliphatic amines e.g.
imipramine.
(ii) Alicyclic amines e.g.
nicotine.
(iii) Nitrogen atoms of aromatic
heterocycles e.g. trimethoprim.
(iv) Amines attached to aromatic rings e.g.
N,N-dimethyl aniline.
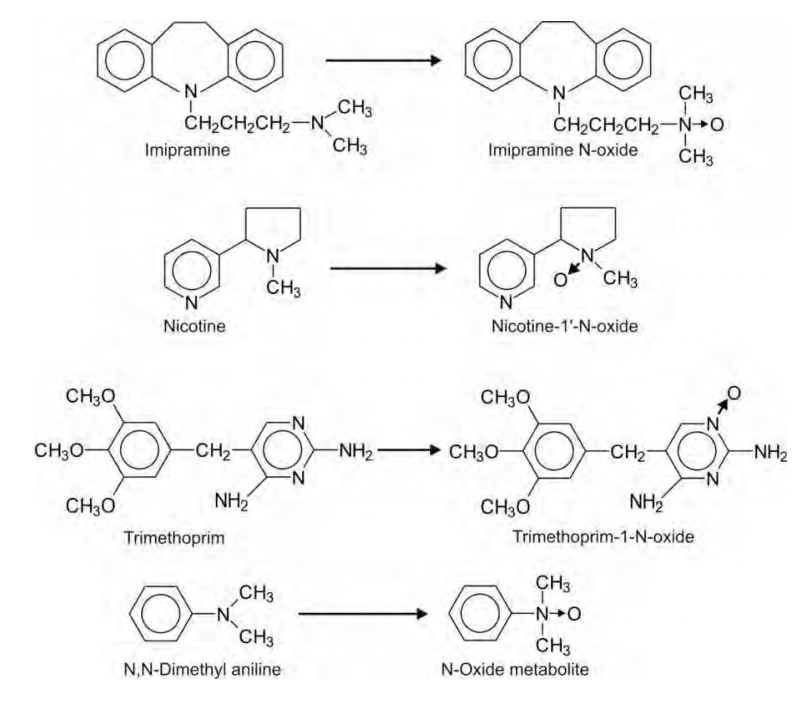
The N-oxide products are highly water-soluble and
excreted in urine. They are, however, susceptible to reduction to the
corresponding amine.
4. N-Hydroxylation: Converse to basic compounds that
form N-oxides, N-hydroxy formation
is usually displayed by non-basic nitrogen atoms such as amide nitrogen, e.g.
lidocaine.
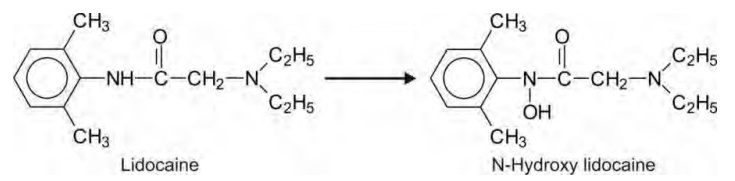
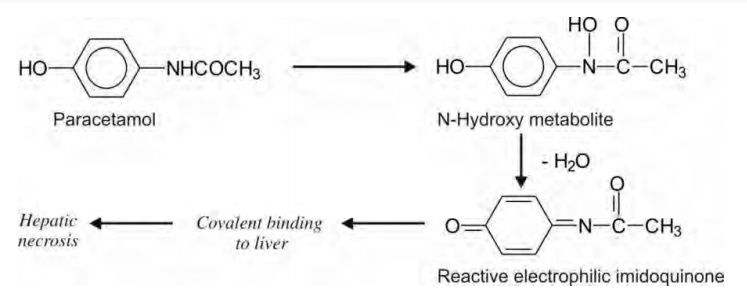
N-hydroxylation of amides often leads to generation
of chemically reactive intermediates capable of binding covalently with
macromolecules, e.g. paracetamol. Paracetamol is safe in therapeutic doses
since its reactive metabolite imidoquinone is neutralized by glutathione.
However, in high doses, the glutathione level becomes insufficient and
significant covalent tissue binding thus occurs resulting in hepatotoxicity.
Phenacetin is also toxic for the same reason.
Non-basic aromatic amines also undergo
hydroxylation. A primary aromatic amine is further converted to nitroso
derivative, e.g. dapsone.
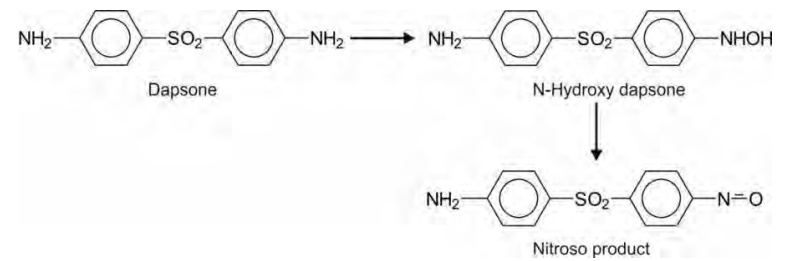
The N-hydroxy dapsone can oxidize ferrous form of
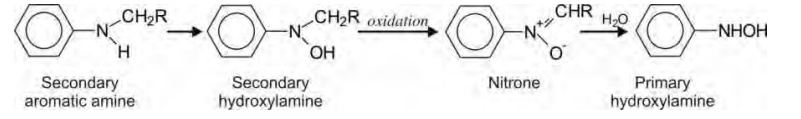
haemoglobin to ferric form and cause methemoglobinaemia. A secondary aromatic
amine yields a nitrone subsequent to formation of secondary hydroxylamine which
is further hydrated to primary hydroxylamine.
N-hydroxylation is also possible at basic nitrogen,
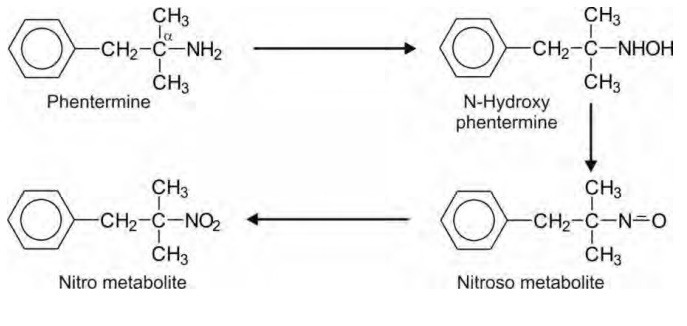
e.g. primary and secondary amines. Phentermine, a primary aliphatic amine,
cannot undergo deamination as the -carbon does not contain hydrogen and the
drug is therefore N-hydroxylated.
Oxidation of Carbon-Sulphur Systems
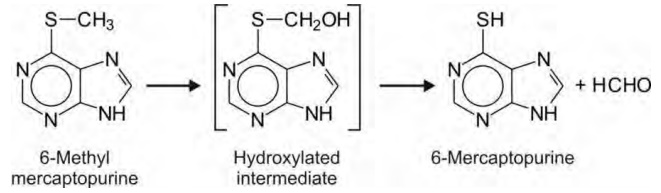
1. S-Dealkylation: The mechanism of S-dealkylation
of thioethers (RSR’) is analogous to N-dealkylation i.e. it proceeds via -carbon hydroxylation. The C-S bond
cleavage results in formation of a thiol (RSH) and a carbonyl product, e.g.
6-methyl mercaptopurine.
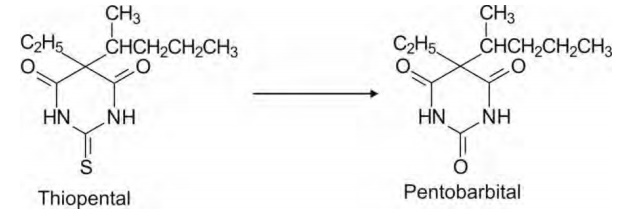
2. Desulphuration: This reaction also involves
cleavage of carbon-sulphur bond (C=S or
thiono). The product is the one with C=O bond. Such a desulphuration reaction
is commonly observed in thioamides (RCSNHR’) such as thiopental.
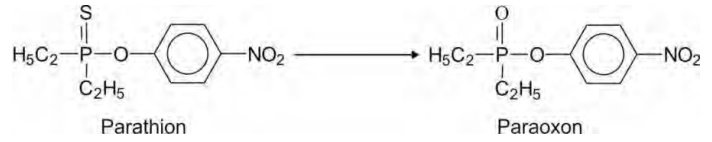
Desulphuration also occurs with compounds
containing P=S bonds such as the organophosphate pesticides, e.g. parathion.
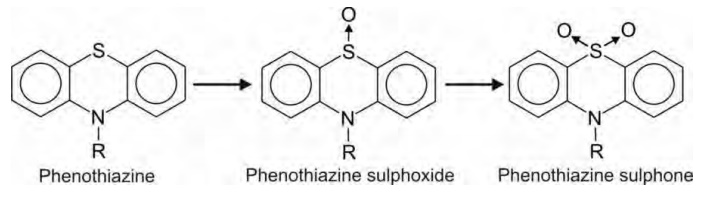
3. S-Oxidation: Apart from S-dealkylation,
thioethers can also undergo S-oxidation reactions
to yield sulphoxides which may be further oxidised to sulphones (RSO2R).
Several phenothiazines, e.g. chlorpromazine, undergo S-oxidation.
Oxidation of Carbon-Oxygen Systems
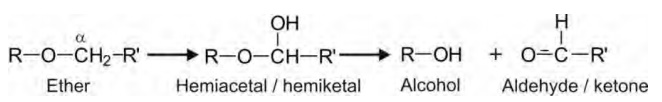
O-Dealkylation: This reaction is also similar to
N-dealkylation and proceeds by α-carbon hydroxylation to form an
unstable hemiacetal or hemiketal intermediate which spontaneously undergoes C-O
bond cleavage to form alcohol (arenol or alkanol) and a carbonyl moiety.
The O-containing functional groups (analogous to
amines and amides, the substrates which undergo N-dealkylation) are ethers and
esters. However, only the ethers undergo O-carbonyl moiety.-dealkylation
reaction. Aliphatic ether drugs are rare and aromatic ethers (phenolic) are
common. Methyl ethers are rapidly dealkylated in comparison to longer chain
ethers such as the one containing n-butyl group. The reaction generally leads
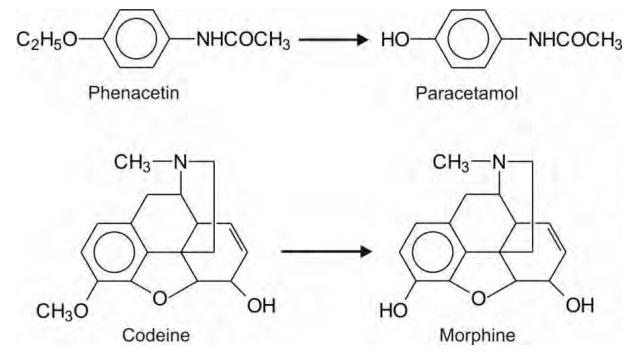
to formation of active metabolites, e.g. phenacetin to paracetamol, and codeine
to morphine.
Oxidation of Alcohol, Carbonyl and Carboxylic Acid
These reactions are mainly catalysed by
non-microsomal enzymes, dehydrogenases. Primary and secondary alcohols and
aldehydes undergo oxidation relatively easily but tertiary alcohols, ketones
and carboxylic acids are resistant since such a reaction involves cleavage of
C-C bonds.
Primary alcohols are rapidly metabolised to
aldehydes (and further to carboxylic acids) but oxidation of secondary alcohols
to ketones proceeds slowly. This is because –
1. Primary alcohols are better
substrates for dehydrogenases due to their acidic nature, and
2. Their oxidation products,
carboxylic acids, are more water-soluble than ketones.
Since primary alcohols are inactivated rapidly,
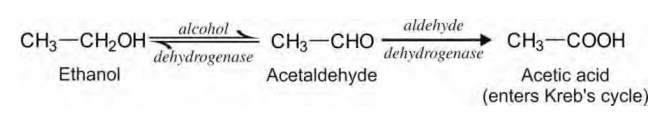
drugs bearing such groups are rare. In case of ethanol, oxidation to
acetaldehyde is reversible and further oxidation of the latter to acetic acid
is very rapid since acetaldehyde is highly toxic and should not accumulate in
the body. Compounds with two primary alcohol functions are oxidised stepwise
and not simultaneously. With secondary alcohols, the rate of oxidation
increases with an increase in alkyl chain length. Compounds with both primary
and secondary alcohol groups are oxidised preferentially at the primary group.
Miscellaneous Oxidative Reactions
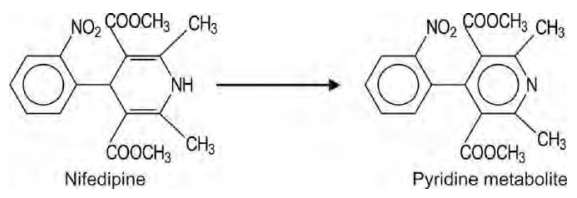
a. Oxidative Aromatisation/Dehydrogenation: An example of metabolic aromatisation of drugs is nifedipine.
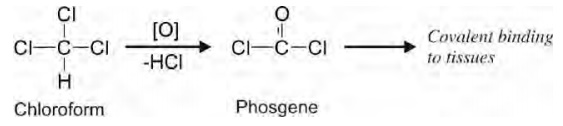
b. Oxidative Dehalogenation: This
reaction is common with halogen containing drugs such as chloroform. Dehalogenation of this drug yields phosgene
which may result in electrophiles capable of covalent binding to tissues.
Oxidative ring cleavage, oxidation of arenols to
quinones, etc. are other oxidative reactions.
Related Topics
