Elastin
| Home | | Biochemistry |Chapter: Biochemistry : Fibrous Proteins
In contrast to collagen, which forms fibers that are tough and have high tensile strength, elastin is a connective tissue protein with rubber-like properties.
ELASTIN
In contrast to
collagen, which forms fibers that are tough and have high tensile strength,
elastin is a connective tissue protein with rubber-like properties. Elastic
fibers composed of elastin and glycoprotein microfibrils are found in the
lungs, the walls of large arteries, and elastic ligaments. They can be stretched
to several times their normal length but recoil to their original shape when
the stretching force is relaxed.
A. Structure
Elastin is an insoluble
protein polymer synthesized from a precursor, tropoelastin, which is a linear
polypeptide composed of about 700 amino acids that are primarily small and
nonpolar (for example, glycine, alanine, and valine). Elastin is also rich in
proline and lysine but contains scant hydroxyproline and hydroxylysine.
Tropoelastin is secreted by the cell into the extracellular space. There, it
interacts with specific glycoprotein microfibrils, such as fibrillin, which
function as a scaffold onto which tropoelastin is deposited. Some of the lysyl
side chains of the tropoelastin polypeptides are oxidatively deaminated by lysyl
oxidase, forming allysine residues. Three of the allysyl side chains plus one
unaltered lysyl side chain from the same or neighboring polypeptides form a
desmosine cross-link (Figure 4.12). This produces elastin, an extensively
interconnected, rubbery network that can stretch and bend in any direction when
stressed, giving connective tissue elasticity (Figure 4.13). Mutations in the
fibrillin-1 protein are responsible for Marfan syndrome, a connective tissue
disorder characterized by impaired structural integrity in the skeleton, the
eye, and the cardiovascular system. With this disease, abnormal fibrillin
protein is incorporated into microfibrils along with normal fibrillin,
inhibiting the formation of functional microfibrils. [Note: Patients with
Marfan syndrome, OI, or EDS may have blue sclerae due to tissue thinning that
allows underlying pigment to show through.]
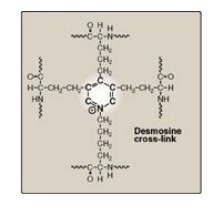
Figure 4.12 Desmosine
cross-link in elastin.
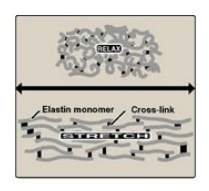
Figure 4.13 Elastin fibers in
relaxed and stretched conformations.
B. Role of α1-antitrypsin in elastin degradation
1. α1-Antitrypsin: Blood and other body fluids
contain a protein, α1-antitrypsin (AAT or A1AT), which inhibits a number of
proteolytic enzymes (called proteases or proteinases) that hydrolyze and
destroy proteins. [Note: The inhibitor was originally named α1-antitrypsin
because it inhibits the activity of trypsin, a proteolytic enzyme synthesized
as trypsinogen by the pancreas.] AAT has the important physiologic role of
inhibiting neutrophil elastase, a powerful protease that is released into the
extracellular space and degrades elastin of alveolar walls as well as other
structural proteins in a variety of tissues (Figure 4.14). Most of the AAT
found in plasma is synthesized and secreted by the liver. AAT comprises more
than 90% of the α1-globulin fraction of normal plasma. Extrahepatic synthesis
occurs in monocytes and alveolar macrophages, and may be important in the
prevention of local tissue injury by elastase.
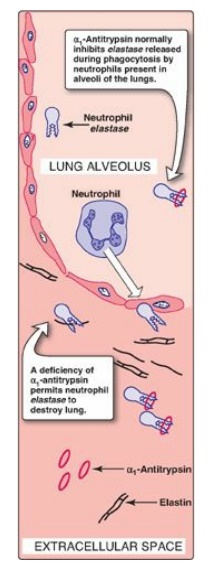
Figure 4.14 Destruction of alveolar tissue by elastase released from neutrophils activated as part of the immune response to airborne pathogens.
2. Role of α1-antitrypsin in the lungs: In the normal lung, the alveoli
are chronically exposed to low levels of neutrophil elastase released from
activated and degenerating neutrophils. The proteolytic activity of elastase
can destroy the elastin in alveolar walls if unopposed by the action of AAT,
the most important inhibitor of neutrophil elastase (see Figure 4.14). Because
lung tissue cannot regenerate, the destruction of the connective tissue of
alveolar walls results in emphysema.
3. Emphysema resulting from α1-antitrypsin
deficiency: In the
United States, approximately 2%–5% of patients with emphysema are predisposed
to the disease by inherited defects in AAT. A number of different mutations in
the gene for AAT are known to cause a deficiency of the protein, but one single
purine base mutation (GAG to AAG, resulting in the substitution of lysine for
glutamic acid at position 342 of the protein) is clinically the most
widespread. The mutation causes the normally monomeric AAT to polymerize within
the endoplasmic reticulum of hepatocytes, resulting in decreased secretion of
AAT by the liver. Consequently, blood levels of AAT are reduced, decreasing the
amount that gets to the alveoli. The polymer that accumulates in the liver may
result in cirrhosis (scarring of the liver). In the United States, the AAT
mutation is most common in Caucasians of Northern European ancestry. An
individual must inherit two abnormal AAT alleles to be at risk for the
development of emphysema. In a heterozygote, with one normal and one defective
gene, the levels of AAT are sufficient to protect the alveoli from damage.
[Note: Methionine 358 in AAT is required for the binding of the inhibitor to
its target proteases. Smoking causes the oxidation and subsequent inactivation
of the methionine, thereby rendering the inhibitor powerless to neutralize
elastase. Smokers with AAT deficiency, therefore, have a considerably elevated
rate of lung destruction and a poorer survival rate than nonsmokers with the
deficiency.] The deficiency of elastase inhibitor can be treated by weekly
augmentation therapy, that is, intravenous administration of AAT. The AAT
diffuses from the blood into the lung, where it reaches therapeutic levels in
the fluid surrounding the lung epithelial cells.
Related Topics
