Hemoglobinopathies
| Home | | Biochemistry |Chapter: Biochemistry : Globular Proteins
Hemoglobinopathies are defined as a group of genetic disorders caused by production of a structurally abnormal hemoglobin molecule; synthesis of insufficient quantities of normal hemoglobin; or, rarely, both.
HEMOGLOBINOPATHIES
Hemoglobinopathies are
defined as a group of genetic disorders caused by production of a structurally
abnormal hemoglobin molecule; synthesis of insufficient quantities of normal
hemoglobin; or, rarely, both. Sickle cell anemia (HbS), hemoglobin C disease
(HbC), hemoglobin SC disease (HbS + HbC = HbSC), and the thalassemias are
representative hemoglobinopathies that can have severe clinical consequences.
The first three conditions result from production of hemoglobin with an altered
amino acid sequence (qualitative hemoglobinopathy), whereas the thalassemias
are caused by decreased production of normal hemoglobin (quantitative
hemoglobinopathy).
A. Sickle cell anemia (hemoglobin S disease)
Sickle cell anemia, the
most common of the RBC sickling diseases, is a genetic disorder of the blood
caused by a single nucleotide substitution in the gene for β-globin. It is the
most common inherited blood disorder in the United States, affecting 50,000
Americans. It occurs primarily in the African American population, affecting
one of 500 newborn African American infants in the United States. Sickle cell
anemia is an autosomal recessive disorder. It occurs in individuals who have
inherited two mutant genes (one from each parent) that code for synthesis of
the β chains of the globin molecules. [Note: The mutant β-globin chain is
designated βS, and the resulting hemoglobin, α2βS2, is
referred to as HbS.] An infant does not begin showing symptoms of the disease
until sufficient HbF has been replaced by HbS so that sickling can occur (see
below). Sickle cell anemia is characterized by lifelong episodes of pain
(“crises”); chronic hemolytic anemia with associated hyperbilirubinemia; and
increased susceptibility to infections, usually beginning in infancy. [Note:
The lifetime of a RBC in sickle cell anemia is less than 20 days, compared with
120 days for normal RBC, hence, the anemia.] Other symptoms include acute chest
syndrome, stroke, splenic and renal dysfunction, and bone changes due to marrow
hyperplasia. Heterozygotes, representing 1 in 12 African Americans, have one
normal and one sickle cell gene. The blood cells of such heterozygotes contain
both HbS and HbA. These individuals have sickle cell trait. They usually do not
show clinical symptoms (but may under conditions of extreme physical exertion
with dehydration) and can have a normal life span.
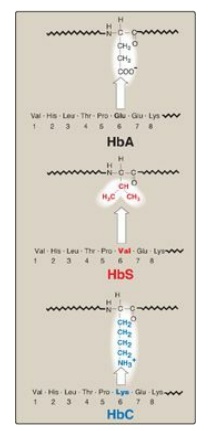
Figure 3.18
Amino acid substitutions in hemoglobin S (HbS) and hemoglobin C (HbC).
1. Amino acid substitution in HbS β chains: A molecule of HbS contains two
normal α-globin chains and two mutant β-globin chains (βS), in which glutamate
at position six has been replaced with valine (Figure 3.18). Therefore, during
electrophoresis at alkaline pH, HbS migrates more slowly toward the anode
(positive electrode) than does HbA (Figure 3.19). This altered mobility of HbS
is a result of the absence of the negatively charged glutamate residues in the
two β chains, thereby rendering HbS less negative than HbA. [Note:
Electrophoresis of hemoglobin obtained from lysed RBC is routinely used in the
diagnosis of sickle cell trait and sickle cell disease. DNA analysis also is
used.]
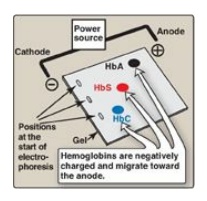
Figure 3.19 Diagram of hemoglobins (HbA), (HbS), and (HbC) after electrophoresis.
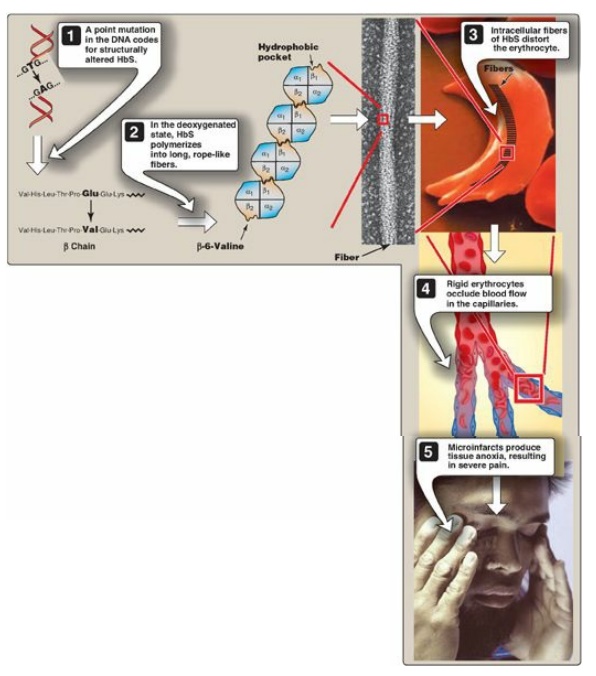
Figure 3.20 Molecular and cellular events leading to sickle cell crisis. HbS = hemoglobin S.
2. Sickling and tissue anoxia: The replacement of the charged
glutamate with the nonpolar valine forms a protrusion on the β chain that fits
into a complementary site on the β chain of another hemoglobin molecule in the
cell (Figure 3.20). At low oxygen tension, deoxyhemoglobin S polymerizes inside
the RBC, forming a network of insoluble fibrous polymers that stiffen and
distort the cell, producing rigid, misshapen RBC. Such sickled cells frequently
block the flow of blood in the narrow capillaries. This interruption in the
supply of oxygen leads to localized anoxia (oxygen deprivation) in the tissue,
causing pain and eventually death (infarction) of cells in the vicinity of the
blockage. The anoxia also leads to an increase in deoxygenated HbS. [Note: The
mean diameter of RBC is 7.5 µm, whereas that of the microvasculature is 3–4 µm.
Compared to normal RBC, sickled cells have a decreased ability to deform and an
increased tendency to adhere to vessel walls and so have difficulty moving
through small vessels, thereby causing microvascular occlusion.]
3. Variables that increase sickling: The extent of sickling and,
therefore, the severity of disease is enhanced by any variable that increases
the proportion of HbS in the deoxy state (that is, reduces the affinity of HbS
for O2). These variables include decreased pO2, increased
pCO2, decreased pH, dehydration, and an increased concentration of
2,3-BPG in RBC.
4. Treatment: Therapy involves adequate hydration, analgesics, aggressive antibiotic therapy if infection is present, and transfusions in patients at high risk for fatal occlusion of blood vessels. Intermittent transfusions with packed RBC reduce the risk of stroke, but the benefits must be weighed against the complications of transfusion, which include iron overload (hemosiderosis), bloodborne infections, and immunologic complications. Hydroxyurea (hydroxycarbamide), an antitumor drug, is therapeutically useful because it increases circulating levels of HbF, which decreases RBC sickling. This leads to decreased frequency of painful crises and reduces mortality. [Note: The morbidity and mortality associated with sickle cell anemia has led to its inclusion in newborn screening panels to allow prophylactic antibiotic therapy to begin soon after the birth of an affected child.]
5. Possible selective advantage of the heterozygous
state: The
high frequency of the bS mutation among black Africans, despite its damaging
effects in the homozygous state, suggests that a selective advantage exists for
heterozygous individuals. For example, heterozygotes for the sickle cell gene
are less susceptible to the severe malaria caused by the parasite Plasmodium
falciparum. This organism spends an obligatory part of its life cycle in the
RBC. One theory is that because these cells in individuals heterozygous for
HbS, like those in homozygotes, have a shorter life span than normal, the
parasite cannot complete the intracellular stage of its development. This fact
may provide a selective advantage to heterozygotes living in regions where
malaria is a major cause of death. Figure 3.21 illustrates that in Africa, the
geographic distribution of sickle cell anemia is similar to that of malaria.
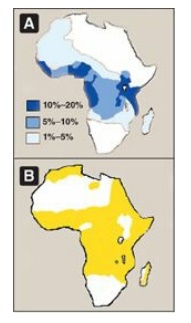
Figure 3.21 A. Distribution of sickle cell in Africa expressed as a percentage of the population with disease. B. Distribution of malaria in Africa.
B. Hemoglobin C disease
Like HbS, HbC is a
hemoglobin variant that has a single amino acid substitution in the sixth
position of the β-globin chain (see Figure 3.18). In HbC, however, a lysine is
substituted for the glutamate (as compared with a valine substitution in HbS).
[Note: This substitution causes HbC to move more slowly toward the anode than
HbA or HbS does (see Figure 3.19).] Rare patients homozygous for HbC generally
have a relatively mild, chronic hemolytic anemia. These patients do not suffer
from infarctive crises, and no specific therapy is required.
C. Hemoglobin SC disease
HbSC disease is another
of the RBC sickling diseases. In this disease, some β-globin chains have the
sickle cell mutation, whereas other β-globin chains carry the mutation found in
HbC disease. [Note: Patients with HbSC disease are doubly heterozygous. They
are called compound heterozygotes because both of their β-globin genes are
abnormal, although different from each other.] Hemoglobin levels tend to be
higher in HbSC disease than in sickle cell anemia and may even be at the low
end of the normal range. The clinical course of adults with HbSC anemia differs
from that of sickle cell anemia in that symptoms such as painful crises are
less frequent and less severe. However, there is significant clinical
variability.
D. Methemoglobinemias
Oxidation of the heme iron in hemoglobin to the ferric (Fe3+) state forms methemoglobin, which cannot bind O2. This oxidation may be caused by the action of certain drugs, such as nitrates, or endogenous products such as reactive oxygen species. The oxidation may also result from inherited defects, for example, certain mutations in the α- or β-globin chain promote the formation of methemoglobin (HbM). Additionally, a deficiency of NADH-cytochrome b5 reductase (also called NADH-methemoglobin reductase), the enzyme responsible for the conversion of methemoglobin (Fe3+) to hemoglobin (Fe2+), leads to the accumulation of HbM. [Note: The RBC of newborns have approximately half the capacity of those of adults to reduce HbM. They are, therefore, particularly susceptible to the effects of HbM-producing compounds.] The methemoglobinemias are characterized by “chocolate cyanosis” (a brownish blue coloration of the skin and mucous membranes and brown-colored blood) as a result of the dark-colored HbM. Symptoms are related to the degree of tissue hypoxia and include anxiety, headache, and dyspnea. In rare cases, coma and death can occur. Treatment is with methylene blue, which is oxidized as Fe+3 is reduced.
E. Thalassemias
The thalassemias are
hereditary hemolytic diseases in which an imbalance occurs in the synthesis of
globin chains. As a group, they are the most common single gene disorders in
humans. Normally, synthesis of the α- and β-globin chains is coordinated, so
that each α-globin chain has a β-globin chain partner. This leads to the
formation of α2β2 (HbA). In the thalassemias, the
synthesis of either the α- or the β-globin chain is defective. A thalassemia can
be caused by a variety of mutations, including entire gene deletions, or
substitutions or deletions of one to many nucleotides in the DNA. [Note: Each
thalassemia can be classified as either a disorder in which no globin chains
are produced (αo- or βo-thalassemia), or one in which
some chains are synthesized but at a reduced level (α+- or β+-thalassemia).]
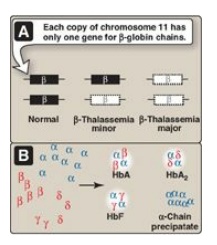
Figure 3.22
A. β-Globin gene mutations in the β-thalassemias. B. Hemoglobin (Hb) tetramers
formed in β-thalassemias.
1. β-Thalassemias: In these disorders, synthesis of
β-globin chains is decreased or absent, typically as a result of point
mutations that affect the production of functional mRNA. However, α-globin
chain synthesis is normal. Excess α-globin chains cannot form stable tetramers
and so precipitate, causing the premature death of cells initially destined to
become mature RBC. Increase in α2δ2 (HbA2) and
α2γ2 (HbF) also occurs. There are only two copies of the β-globin
gene in each cell (one on each chromosome 11). Therefore, individuals with
β-globin gene defects have either β-thalassemia trait (β-thalassemia minor) if
they have only one defective β-globin gene or β-thalassemia major (Cooley
anemia) if both genes are defective (Figure 3.22). Because the β-globin gene is
not expressed until late in fetal gestation, the physical manifestations of
β-thalassemias appear only several months after birth. Those individuals with
β-thalassemia minor make some β chains, and usually do not require specific
treatment. However, those infants born with β-thalassemia major are seemingly
healthy at birth but become severely anemic, usually during the first or second
year of life due to ineffective erythropoiesis. Skeletal changes as a result of
extramedullary hematopoiesis also are seen. These patients require regular
transfusions of blood. [Note: Although this treatment is lifesaving, the
cumulative effect of the transfusions is iron overload (a syndrome known as
hemosiderosis). Use of iron chelation therapy has improved morbidity and
mortality.] The only curative option available is hematopoietic stem cell
transplantation.
2. α-Thalassemias: In these disorders, synthesis of
α-globin chains is decreased or absent, typically as a result of deletional
mutations. Because each individual’s genome contains four copies of the
α-globin gene (two on each chromosome 16), there are several levels of α-globin
chain deficiencies (Figure 3.23). If one of the four genes is defective, the
individual is termed a silent carrier of α-thalassemia, because no physical
manifestations of the disease occur. If two α-globin genes are defective, the individual
is designated as having α-thalassemia trait. If three α-globin genes are
defective, the individual has hemoglobin H (β4) disease, a hemolytic
anemia of variable severity. If all four α-globin genes are defective,
hemoglobin Bart (γ4) disease with hydrops fetalis and fetal death
results, because α-globin chains are required for the synthesis of HbF.
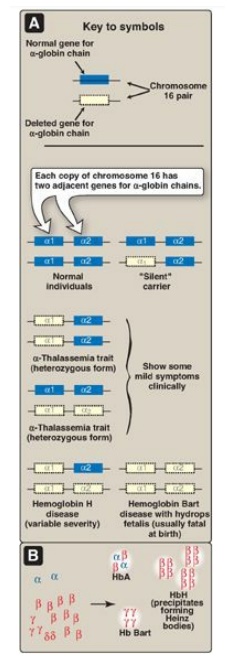
Figure 3.23
A. α-Globin gene deletions in the α-thalassemias. B. Hemoglobin (Hb) tetramers
formed in α-thalassemias.
