Globular Hemeproteins
| Home | | Biochemistry |Chapter: Biochemistry : Globular Proteins
Hemeproteins are a group of specialized proteins that contain heme as a tightly bound prosthetic group. (See: for a discussion of prosthetic groups.)
GLOBULAR HEMEPROTEINS
Hemeproteins are a group of specialized proteins that contain heme as a tightly bound prosthetic group. (See: for a discussion of prosthetic groups.) The role of the heme group is dictated by the environment created by the three-dimensional structure of the protein. For example, the heme group of a cytochrome functions as an electron carrier that is alternately oxidized and reduced. In contrast, the heme group of the enzyme catalase is part of the active site of the enzyme that catalyzes the breakdown of hydrogen peroxide. In hemoglobin and myoglobin, the two most abundant hemeproteins in humans, the heme group serves to reversibly bind oxygen.
A. Structure of heme
Heme is a complex of
protoporphyrin IX and ferrous iron (Fe2+) (Figure 3.1). The iron is held in the
center of the heme molecule by bonds to the four nitrogens of the porphyrin
ring. The heme Fe2+ can form two additional bonds, one on each side
of the planar porphyrin ring. In myoglobin and hemoglobin, one of these
positions is coordinated to the side chain of a histidine residue of the globin
molecule, whereas the other position is available to bind oxygen (Figure 3.2).

Figure 3.1 A. Hemeprotein (cytochrome c). B. Structure of heme.
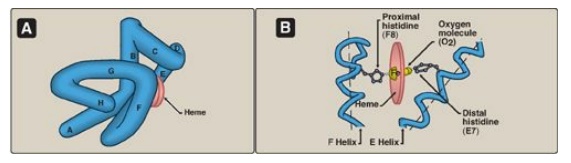
Figure 3.2 A.
Model of myoglobin showing helices A to H. B. Schematic diagram of the
oxygen-binding site of myoglobin.
B. Structure and function of myoglobin
Myoglobin, a
hemeprotein present in heart and skeletal muscle, functions both as a reservoir
for oxygen and as an oxygen carrier that increases the rate of transport of
oxygen within the muscle cell. [Note: Mouse myoglobin double knockouts have,
surprisingly, an apparently normal phenotype.] Myoglobin consists of a single
polypeptide chain that is structurally similar to the individual polypeptide
chains of the tetrameric hemoglobin molecule. This homology makes myoglobin a
useful model for interpreting some of the more complex properties of
hemoglobin.
1. α-Helical content: Myoglobin is a compact molecule,
with approximately 80% of its polypeptide chain folded into eight stretches of
α-helix. These α-helical regions, labeled A to H in Figure 3.2A, are terminated
either by the presence of proline, whose five-membered ring cannot be
accommodated in an α-helix or by β-bends and loops stabilized by hydrogen bonds
and ionic bonds. [Note: Ionic bonds are also termed electrostatic interactions
or salt bridges.]
2. Location of polar and nonpolar amino acid
residues: The
interior of the myoglobin molecule is composed almost entirely of nonpolar
amino acids. They are packed closely together, forming a structure stabilized
by hydrophobic interactions between these clustered residues. In contrast,
polar amino acids are located almost exclusively on the surface, where they can
form hydrogen bonds, both with each other and with water.
3. Binding of the heme group: The heme group of the myoglobin molecule sits in a crevice, which is lined with nonpolar amino acids. Notable exceptions are two histidine residues (Figure 3.2B). One, the proximal histidine (F8), binds directly to the iron of heme. The second, or distal histidine (E7), does not directly interact with the heme group but helps stabilize the binding of oxygen to the ferrous iron. The protein, or globin, portion of myoglobin thus creates a special microenvironment for the heme that permits the reversible binding of one oxygen molecule (oxygenation). The simultaneous loss of electrons by the ferrous iron (oxidation to the ferric form) occurs only rarely.
C. Structure and function of hemoglobin
Hemoglobin is found
exclusively in red blood cells (RBC), where its main function is to transport
oxygen (O2) from the lungs to the capillaries of the tissues.
Hemoglobin A, the major hemoglobin in adults, is composed of four polypeptide
chains (two α chains and two β chains) held together by noncovalent
interactions (Figure 3.3). Each chain (subunit) has stretches of α-helical
structure and a hydrophobic heme-binding pocket similar to that described for
myoglobin. However, the tetrameric hemoglobin molecule is structurally and
functionally more complex than myoglobin. For example, hemoglobin can transport
H+ and CO2 from the tissues to the lungs and can carry four
molecules of O2 from the lungs to the cells of the body.
Furthermore, the oxygen-binding properties of hemoglobin are regulated by
interaction with allosteric effectors.
Obtaining O2 from the atmosphere solely by diffusion greatly limits the size of organisms. Circulatory systems overcome this, but transport molecules such as hemoglobin are also required because O2 is only slightly soluble in aqueous solutions such as blood.
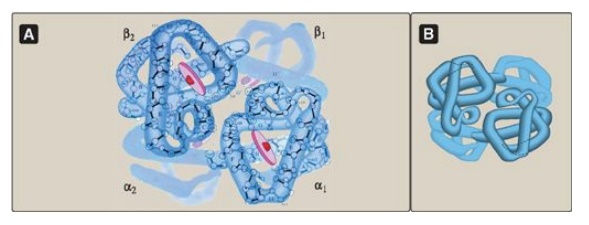
Figure 3.3 A. Structure of hemoglobin showing the polypeptide backbone. B. Simplified drawing showing the helices.
1. Quaternary structure of hemoglobin: The hemoglobin tetramer can be
envisioned as being composed of two identical dimers, (αβ)1 and (αβ)2.
The two polypeptide chains within each dimer are held tightly together
primarily by hydrophobic interactions (Figure 3.4). [Note: In this instance,
hydrophobic amino acid residues are localized not only in the interior of the
molecule, but also in a region on the surface of each subunit. Multiple
interchain hydrophobic interactions form strong associations between α-subunits
and β-subunits in the dimers.] In contrast, the two dimers are held together
primarily by polar bonds. The weaker interactions between the dimers allows
them to move with respect to one other. This movement results in the two dimers
occupying different relative positions in deoxyhemoglobin as compared with
oxyhemoglobin (see Figure 3.4). [Note: The binding of O2 to the heme
iron pulls the iron into the plane of the heme. Because the iron is also linked
to the proximal histidine (F8), there is movement of the globin chains that
alters the interface between the αβ dimers.]
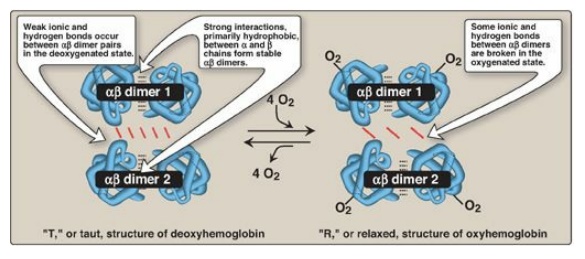
Figure 3.4
Schematic diagram showing structural changes resulting from oxygenation and
deoxygenation of hemoglobin.
a. T form: The deoxy form of hemoglobin is called the “T,” or
taut (tense) form. In the T form, the two αβ dimers interact through a network
of ionic bonds and hydrogen bonds that constrain the movement of the
polypeptide chains. The T conformation is the low-oxygen-affinity form of
hemoglobin.
b. R form: The binding of O2 to hemoglobin causes
the rupture of some of the polar bonds between the αβ dimers, allowing
movement. This leads to a structure called the “R,” or relaxed form (see Figure
3.4). The R conformation is the high-oxygen-affinity form of hemoglobin.
D. Binding of oxygen to myoglobin and hemoglobin
Myoglobin can bind only one molecule of O2, because it contains only one heme group. In contrast, hemoglobin can bind four O2 molecules, one at each of its four heme groups. The degree of saturation (Y) of these oxygen-binding sites on all myoglobin or hemoglobin molecules can vary between zero (all sites are empty) and 100% (all sites are full), as shown in Figure 3.5. [Note: Pulse oximetry is a noninvasive, indirect method of measuring the O2 saturation of arterial blood based on differences in light absorption by oxyhemoglobin and deoxyhemoglobin.]
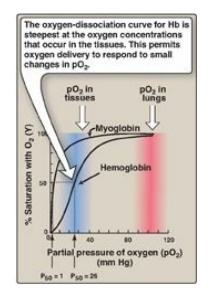
Figure 3.5 Oxygen-dissociation curves for myoglobin and hemoglobin (Hb).
1. Oxygen-dissociation curve: A plot of Y measured at different partial pressures of oxygen (pO2) is called the oxygen-dissociation curve. [Note: pO2 may also be represented as PO2.] The curves for myoglobin and hemoglobin show important differences (see Figure 3.5). This graph illustrates that myoglobin has a higher oxygen affinity at all pO2 values than does hemoglobin. The partial pressure of oxygen needed to achieve half-saturation of the binding sites (P50) is approximately 1 mm Hg for myoglobin and 26 mm Hg for hemoglobin. The higher the oxygen affinity (that is, the more tightly oxygen binds), the lower the P50.
a. Myoglobin: The oxygen-dissociation curve for myoglobin has a
hyperbolic shape (see Figure 3.5). This reflects the fact that myoglobin
reversibly binds a single molecule of oxygen. Thus, oxygenated (MbO2)
and deoxygenated (Mb) myoglobin exist in a simple equilibrium:
Mb + O2 ↔ MbO2
The equilibrium is
shifted to the right or to the left as oxygen is added to or removed from the
system. [Note: Myoglobin is designed to bind oxygen released by hemoglobin at the
low pO2 found in muscle. Myoglobin, in turn, releases oxygen within the muscle
cell in response to oxygen demand.]
b. Hemoglobin: The oxygen-dissociation curve for hemoglobin is sigmoidal in shape (see Figure 3.5), indicating that the subunits cooperate in binding oxygen. Cooperative binding of oxygen by the four subunits of hemoglobin means that the binding of an oxygen molecule at one heme group increases the oxygen affinity of the remaining heme groups in the same hemoglobin tetramer (Figure 3.6). This effect is referred to as heme–heme interaction (see below). Although it is more difficult for the first oxygen molecule to bind to hemoglobin, the subsequent binding of oxygen occurs with high affinity, as shown by the steep upward curve in the region near 20–30 mm Hg (see Figure 3.5).
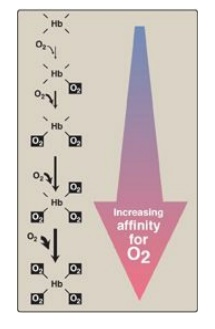
Figure 3.6 Hemoglobin (Hb) binds successive molecules of oxygen with increasing affinity.
E. Allosteric effects
The ability of
hemoglobin to reversibly bind oxygen is affected by the pO2 (through
heme–heme interactions as described above), the pH of the environment, the
partial pressure of carbon dioxide (pCO2) and the availability of
2,3-bisphosphoglycerate. These are collectively called allosteric (“other
site”) effectors, because their interaction at one site on the hemoglobin
molecule affects the binding of oxygen to heme groups at other sites on the
molecule. [Note: The binding of oxygen to monomeric myoglobin is not influenced
by allosteric effectors.]
1. Heme–heme interactions: The sigmoidal oxygen-dissociation curve reflects specific structural changes that are initiated at one heme group and transmitted to other heme groups in the hemoglobin tetramer. The net effect is that the affinity of hemoglobin for the last oxygen bound is approximately 300 times greater than its affinity for the first oxygen bound.
a. Loading and unloading oxygen: The cooperative binding of oxygen
allows hemoglobin to deliver more oxygen to the tissues in response to
relatively small changes in the partial pressure of oxygen. This can be seen in
Figure 3.5, which indicates pO2 in the alveoli of the lung and the
capillaries of the tissues. For example, in the lung, the concentration of
oxygen is high, and hemoglobin becomes virtually saturated (or “loaded”) with
oxygen. In contrast, in the peripheral tissues, oxyhemoglobin releases (or
“unloads”) much of its oxygen for use in the oxidative metabolism of the
tissues (Figure 3.7).
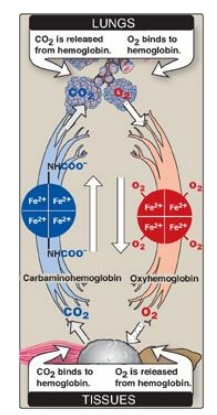
Figure 3.7 Transport of oxygen and carbon dioxide by hemoglobin. Fe = iron.
b. Significance of the sigmoidal oxygen-dissociation curve: The steep slope of the oxygen-dissociation curve over the range of oxygen concentrations that occur between the lungs and the tissues permits hemoglobin to carry and deliver oxygen efficiently from sites of high to sites of low pO2. A molecule with a hyperbolic oxygen-dissociation curve, such as myoglobin, could not achieve the same degree of oxygen release within this range of partial pressures of oxygen. Instead, it would have maximum affinity for oxygen throughout this oxygen pressure range and, therefore, would deliver no oxygen to the tissues.
2. Bohr effect: The release of oxygen from hemoglobin is enhanced
when the pH is lowered or when the hemoglobin is in the presence of an increased
pCO2. Both result in a decreased oxygen affinity of hemoglobin and,
therefore, a shift to the right in the oxygen-dissociation curve (Figure 3.8),
and both, then, stabilize the T (deoxy) form. This change in oxygen binding is
called the Bohr effect. Conversely, raising the pH or lowering the
concentration of CO2 results in a greater affinity for oxygen, a
shift to the left in the oxygen-dissociation curve, and stabilization of the R
(oxy) form.
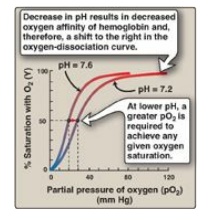
Figure 3.8 Effect of pH on the oxygen affinity of hemoglobin. Protons are allosteric effectors of hemoglobin.
a. Source of the protons that lower the pH: The concentration of both H+ and CO2
in the capillaries of metabolically active tissues is higher than that observed
in alveolar capillaries of the lungs, where CO2 is released into the
expired air. In the tissues, CO2 is converted by carbonic anhydrase
to carbonic acid:
CO2 + H2O
↔ H2CO3
which spontaneously
loses a proton, becoming bicarbonate (the major blood buffer):
H2CO3
↔ HCO3– + H+
The H+ produced by this pair of reactions contributes to the lowering of pH. This differential pH gradient (that is, lungs having a higher pH and tissues a lower pH) favors the unloading of oxygen in the peripheral tissues and the loading of oxygen in the lung. Thus, the oxygen affinity of the hemoglobin molecule responds to small shifts in pH between the lungs and oxygen-consuming tissues, making hemoglobin a more efficient transporter of oxygen.
b. Mechanism of the Bohr effect: The Bohr effect reflects the fact
that the deoxy form of hemoglobin has a greater affinity for protons than does
oxyhemoglobin. This effect is caused by ionizable groups such as specific
histidine side chains that have a higher pKa in deoxyhemoglobin than in
oxyhemoglobin. Therefore, an increase in the concentration of protons
(resulting in a decrease in pH) causes these groups to become protonated
(charged) and able to form ionic bonds (salt bridges). These bonds
preferentially stabilize the deoxy form of hemoglobin, producing a decrease in
oxygen affinity. [Note: Hemoglobin, then, is an important blood buffer.]
The Bohr effect can be
represented schematically as:

where an increase in protons (or a lower pO2) shifts the equilibrium to the right (favoring deoxyhemoglobin), whereas an increase in pO2 (or a decrease in protons) shifts the equilibrium to the left.
3. Effect of 2,3-bisphosphoglycerate on oxygen
affinity:
2,3-Bisphosphoglycerate (2,3-BPG) is an important regulator of the binding of
oxygen to hemoglobin. It is the most abundant organic phosphate in the RBC,
where its concentration is approximately that of hemoglobin. 2,3-BPG is
synthesized from an intermediate of the glycolytic pathway (Figure 3.9; for a
discussion of 2,3-BPG synthesis in glycolysis).
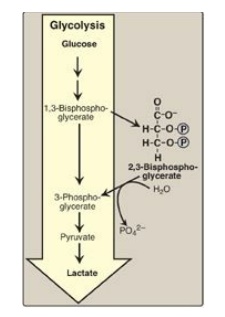
Figure 3.9 Synthesis of 2,3-bisphosphoglycerate. [Note:P is a phosphoryl group, PO32–.] In older literature, 2, 3-bisphosphoglycerate (2,3-BPG) may be referred to as 2,3-diphosphoglycerate (2,3-DPG).
a. Binding of 2,3-BPG to deoxyhemoglobin: 2,3-BPG decreases the O2
affinity of hemoglobin by binding to deoxyhemoglobin but not to oxyhemoglobin.
This preferential binding stabilizes the T conformation of deoxyhemoglobin. The
effect of binding 2,3-BPG can be represented schematically as:

b. Binding site of 2,3-BPG: One molecule of 2,3-BPG binds to a
pocket, formed by the two β-globin chains, in the center of the deoxyhemoglobin
tetramer (Figure 3.10). This pocket contains several positively charged amino
acids that form ionic bonds with the negatively charged phosphate groups of
2,3-BPG. [Note: Replacement of one of these amino acids can result in
hemoglobin variants with abnormally high oxygen affinity that may be
compensated for by increased RBC production (erythrocytosis).] 2,3-BPG is
expelled with oxygenation of the hemoglobin.
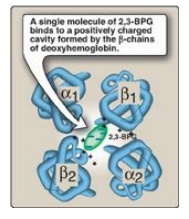
Figure 3.10 Binding of 2,3-bisphosphoglycerate (2,3-BPG) by deoxyhemoglobin.
c. Shift of the oxygen-dissociation curve: Hemoglobin from which 2,3-BPG has been removed has a high affinity for oxygen. However, as seen in the RBC, the presence of 2,3-BPG significantly reduces the affinity of hemoglobin for oxygen, shifting the oxygen-dissociation curve to the right (Figure 3.11). This reduced affinity enables hemoglobin to release oxygen efficiently at the partial pressures found in the tissues.
d. Response of 2,3-BPG levels to chronic hypoxia or
anemia: The
concentration of 2,3-BPG in the RBC increases in response to chronic hypoxia,
such as that observed in chronic obstructive pulmonary disease (COPD) like
emphysema, or at high altitudes, where circulating hemoglobin may have
difficulty receiving sufficient oxygen. Intracellular levels of 2,3-BPG are
also elevated in chronic anemia, in which fewer than normal RBCs are available
to supply the body’s oxygen needs. Elevated 2,3-BPG levels lower the oxygen
affinity of hemoglobin, permitting greater unloading of oxygen in the
capillaries of the tissues (see Figure 3.11).
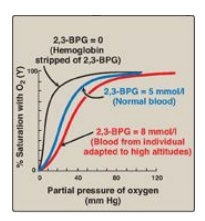
Figure 3.11
Allosteric effect of 2,3-bisphosphoglycerate (2,3-BPG) on the oxygen affinity
of hemoglobin.
e. Role of 2,3-BPG in transfused blood: 2,3-BPG is essential for the
normal oxygen transport function of hemoglobin. However, storing blood in the
currently available media results in a decrease in 2,3-BPG. Stored blood
displays an abnormally high oxygen affinity and fails to unload its bound
oxygen properly in the tissues. Hemoglobin deficient in 2,3-BPG thus acts as an
oxygen “trap” rather than as an oxygen transport system. Transfused RBC are
able to restore their depleted supplies of 2,3-BPG in 6–24 hours. However,
severely ill patients may be compromised if transfused with large quantities of
such 2,3-BPG–“stripped” blood. [Note: The maximum storage time for RBC has been
doubled (21 to 42 days, with median time of 15 days) by changes in H+,
phosphate, and hexose sugar concentration and by the addition of adenine.
Although the content of 2,3-BPG was not greatly improved in the long-term by
these changes, adenosine triphosphate production was increased and improved RBC
survival.]
4. Binding of CO2: Most of the CO2 produced
in metabolism is hydrated and transported as bicarbonate ion. However, some CO
2 is carried as carbamate bound to the N-terminal amino groups of hemoglobin
(forming carbaminohemoglobin as shown in Figure 3.7), which can be represented
schematically as follows:
Hb – NH2 + CO2
↔ Hb – NH – COO- + H+
The binding of CO2
stabilizes the T or deoxy form of hemoglobin, resulting in a decrease in its
affinity for oxygen and a right shift in the oxygen-dissociation curve. In the
lungs, CO2 dissociates from the hemoglobin and is released in the
breath.
5. Binding of CO: Carbon monoxide (CO) binds tightly (but
reversibly) to the hemoglobin iron, forming carboxyhemoglobin. When CO binds to
one or more of the four heme sites, hemoglobin shifts to the R conformation,
causing the remaining heme sites to bind oxygen with high affinity. This shifts
the oxygen-dissociation curve to the left and changes the normal sigmoidal
shape toward a hyperbola. As a result, the affected hemoglobin is unable to
release oxygen to the tissues (Figure 3.12). [Note: The affinity of hemoglobin
for CO is 220 times greater than for oxygen. Consequently, even minute
concentrations of CO in the environment can produce toxic concentrations of
carboxyhemoglobin in the blood. For example, increased levels of CO are found in
the blood of tobacco smokers. CO toxicity appears to result from a combination
of tissue hypoxia and direct CO-mediated damage at the cellular level.] CO
poisoning is treated with 100% oxygen at high pressure (hyperbaric oxygen
therapy), which facilitates the dissociation of CO from the hemoglobin. [Note:
CO inhibits Complex IV of the electron transport chain.] In addition to O2,
CO2, and CO, nitric oxide gas (NO) also is carried by hemoglobin. NO
is a potent vasodilator. It can be taken up (salvaged) or released from RBC,
thus modulating NO availability and influencing vessel diameter.
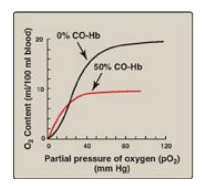
Figure 3.12 Effect
of carbon monoxide (CO) on the oxygen affinity of hemoglobin. CO-Hb =
carboxyhemoglobin (carbon monoxyhemoglobin).
F. Minor hemoglobins
It is important to
remember that human hemoglobin A (HbA) is just one member of a functionally and
structurally related family of proteins, the hemoglobins (Figure 3.13). Each of
these oxygen-carrying proteins is a tetramer, composed of two α-globin (or
α-like) polypeptides and two β-globin (or β-like) polypeptides. Certain
hemoglobins, such as HbF, are normally synthesized only during fetal
development, whereas others, such as HbA2, are synthesized in the adult,
although at low levels compared with HbA. HbA can also become modified by the
covalent addition of a hexose.
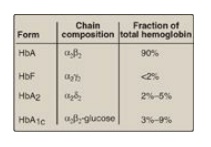
Figure 3.13 Normal adult human hemoglobins. [Note: The α-chains in these hemoglobins are identical.] Hb = hemoglobin.
1. Fetal hemoglobin: HbF is a tetramer consisting of
two α chains identical to those found in HbA, plus two γ chains (α2γ2;
see Figure 3.13). The γ chains are members of the β-globin gene family.
a. HbF synthesis during development: In the first month after
conception, embryonic hemoglobins such as Hb Gower 1, composed of two α-like
zeta (ζ) chains and two β-like epsilon (ε) chains (ζ2ε2),
are synthesized by the embryonic yolk sac. In the fifth week of gestation, the
site of globin synthesis shifts, first to the liver and then to the marrow, and
the primary product is HbF. HbF is the major hemoglobin found in the fetus and
newborn, accounting for about 60% of the total hemoglobin in the RBC during the
last months of fetal life (Figure 3.14). HbA synthesis starts in the bone
marrow at about the eighth month of pregnancy and gradually replaces HbF. (
Figure 3.14 shows the relative production of each type of hemoglobin chain
during fetal and postnatal life.) [Note: HbF represents less than 1% of the
hemoglobin in most adults and is concentrated in RBC known as F cells.]
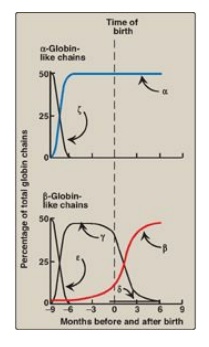
Figure 3.14 Developmental changes in hemoglobin.
b. Binding of 2,3-BPG to HbF: Under physiologic conditions, HbF has a higher affinity for oxygen than does HbA as a result of HbF only weakly binding 2,3-BPG. [Note: The γ-globin chains of HbF lack some of the positively charged amino acids that are responsible for binding 2,3-BPG in the β-globin chains.] Because 2,3-BPG serves to reduce the affinity of hemoglobin for oxygen, the weaker interaction between 2,3-BPG and HbF results in a higher oxygen affinity for HbF relative to HbA. In contrast, if both HbA and HbF are stripped of their 2,3-BPG, they then have a similar affinity for oxygen. The higher oxygen affinity of HbF facilitates the transfer of oxygen from the maternal circulation across the placenta to the RBC of the fetus.
2. Hemoglobin A2: HbA2 is a minor
component of normal adult hemoglobin, first appearing shortly before birth and,
ultimately, constituting about 2% of the total hemoglobin. It is composed of
two α-globin chains and two δ-globin chains (α2δ2; see
Figure 3.13).
3. Hemoglobin A1c: Under physiologic conditions, HbA
is slowly and nonenzymically glycosylated (glycated), the extent of
glycosylation being dependent on the plasma concentration of a particular
hexose. The most abundant form of glycosylated hemoglobin is HbA1c.
It has glucose residues attached predominantly to the NH2 groups of
the N-terminal valines of the β-globin chains (Figure 3.15). Increased amounts
of HbA1c are found in RBC of patients with diabetes mellitus, because their HbA
has contact with higher glucose concentrations during the 120-day lifetime of
these cells. (See : for a discussion of the use of HbA1c levels in assessing
average blood glucose levels in patients with diabetes.)
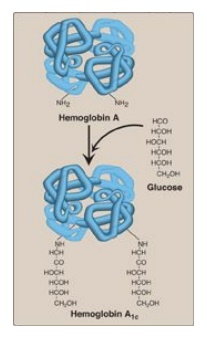
Figure 3.15 Nonenzymic
addition of glucose to hemoglobin. The nonenzymic addition of a sugar to a
protein is referred to as glycation.
Related Topics
