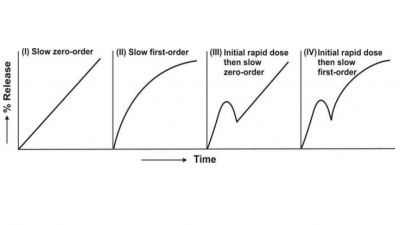
Factors in the Design of Controlled-Release Drug Delivery Systems
| Home | | Biopharmaceutics and Pharmacokinetics |Chapter: Biopharmaceutics and Pharmacokinetics : Controlled Release Medication
A. Biopharmaceutic Characteristics of a Drug in the Design of CRDDS B. Pharmacokinetic Characteristics of a Drug in the Design of CRDDS C. Pharmacodynamic Characteristics of a Drug in the Design of CRDDS
FACTORS IN THE DESIGN OF CONTROLLED-RELEASE DRUG DELIVERY SYSTEMS
The basic rationale
of a controlled release drug delivery system is to optimise the
biopharmaceutic, pharmacokinetic and pharmacodynamic properties of a drug in
such a way that its utility is maximized through reduction in side effects and
cure or control of condition in the shortest possible time by using smallest
quantity of drug, administered by the most suitable route.
A. Biopharmaceutic Characteristics of a Drug in the Design of CRDDS
The performance of a drug presented as a
controlled-release system depends upon its:
1. Release from the formulation.
2. Movement within the body
during its passage to the site of action.
The former depends upon the fabrication of the
formulation and the physicochemical properties of the drug while the latter
element is dependent upon pharmacokinetics of drug. In comparison to
conventional dosage form where the rate-limiting step in drug availability is
usually absorption through the biomembrane, the rate-determining step in the
availability of a drug from controlled delivery system is the rate of release
of drug from the dosage form which is much smaller than the intrinsic
absorption rate for the drug (Fig. 14.2).
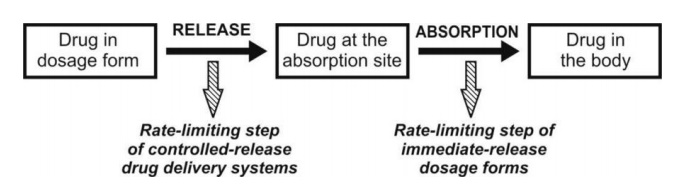
Fig. 14.2 Scheme representing the rate-limiting step in the design of
controlled-release drug delivery
system
The type of delivery system and the route of
administration of the drug presented in controlled-release dosage form depend
upon the physicochemical properties of the drug and its biopharmaceutic
characteristics. The desired biopharmaceutic properties of a drug to be used in
a controlled-release drug delivery system are discussed below.
1. Molecular Weight of the Drug: Lower the
molecular weight, faster and more complete
the absorption. For drugs absorbed by pore transport mechanism, the molecular
size threshold is 150 Daltons for spherical compounds and 400 Daltons for
linear compounds. However, more than 95% of drugs are absorbed by passive
diffusion. Diffusivity, defined
as the ability of a drug to diffuse through the membranes, is inversely related to molecular size.
The upper limit of drug molecular size for passive diffusion is 600 Daltons.
Drugs with large molecular size are poor candidates for oral controlled-release
systems e.g. peptides and proteins.
2. Aqueous Solubility of the Drug: A drug
with good aqueous solubility, especially
if pH-independent, serves as a good candidate for controlled-release dosage
forms e.g. pentoxifylline. The lower limit of solubility of a drug to be
formulated as CRDDS is 0.1mg/ml. Drugs with pH-dependent aqueous solubility
e.g. phenytoin, or drugs with solubility in non-aqueous solvents e.g. steroids,
are suitable for parenteral (e.g. i.m depots) controlled-release dosage forms;
the drug precipitates at the injection site and thus, its release is slowed
down due to change in pH or contact with aqueous body fluids. Solubility of
drug can limit the choice of mechanism to be employed in CRDDS, for example,
diffusional systems are not suitable for poorly soluble drugs. Absorption of
poorly soluble drugs is dissolution rate-limited which means that the
controlled-release device does not control the absorption process; hence, they
are poor candidates for such systems.
3. Apparent Partition Coefficient/Lipophilicity of the Drug: Greater the apparent partition
coefficient of a drug, greater its lipophilicity and thus, greater is its rate
and extent of absorption. Such drugs have increased tendency to cross even the
more selective barriers like BBB. The apparent volume of distribution of such
drugs also increases due to increased partitioning into the fatty tissues and
since the blood flow rate to such tissues is always lower than that to an
aqueous tissue like liver, they may exhibit characteristics of models having
two or more compartments. The parameter is also important in determining the
release rate of a drug from lipophilic matrix or device.
4. Drug pKa and Ionisation at Physiological pH: The pKa range for
acidic drugs whose ionisation is
pH-sensitive is 3.0 to 7.5 and that for basic drugs is 7.0 to 11.0. For optimum
passive absorption, the drugs should be non-ionised at that site at least to an
extent 0.1 to 5%. Drugs existing largely in ionised forms are poor candidates
for controlled delivery e.g. hexamethonium.
5. Drug Permeability: The three major drug characteristics that determine the permeability
of drugs for passive transport across intestinal epithelium are –
·
Lipophilicity, expressed as log
P.
·
Polarity of drug which is
measured by the number of H-bond acceptors and number of H-bond donors on the
drug molecule.
·
Molecular size.
The influence of each of these properties has been
discussed above.
6. Drug Stability: Drugs unstable in GI environment cannot be
administered as oral controlled-release
formulation because of bioavailability problems e.g. nitroglycerine. A
different route of administration should then be selected such as the
transdermal route. Drugs unstable in gastric pH, e.g. propantheline can be
designed for sustained delivery in intestine with limited or no delivery in
stomach. On the other hand, a drug unstable in intestine, e.g. probanthine, can
be formulated as gastroretentive dosage form.
7. Mechanism and Site of Absorption: Drugs absorbed
by carrier-mediated transport
processes and those absorbed through a window
are poor candidates for controlled-release systems e.g. several B vitamins.
8. Biopharmaceutic Aspects of Route of Administration: Oral and parenteral (i.m.)
routes are the most popular followed by transdermal application. Routes of
minor importance in controlled drug delivery are buccal/sublingual, rectal,
nasal, ocular, pulmonary, vaginal and intrauterinal. The features desirable for
a drug to be given by a particular route are discussed below.
(a) Oral Route: For a drug to be successful as oral
controlled-release formulation, it must
get absorbed through the entire length of GIT. Since the main limitation of
this route is the transit time (a mean of 14 hours), the duration of action can
be extended for 12 to 24 hours. The route is suitable for drugs given in dose
as high as 1000 mg. A drug, whose absorption is pH-dependent, destabilized by
GI fluids/enzymes, undergoes extensive presystemic metabolism (e.g.
nitroglycerine), influenced by gut motility, has an absorption window and/or
absorbed actively (e.g. riboflavin), is a poor candidate for oral
controlled-release formulations.
(b) Intramuscular/Subcutaneous Routes: These routes are suitable when the
duration of action is to be prolonged from 24 hours to 12 months. Only a
small amount of drug, about 2 ml or 2 grams, can be administered by these
routes. Factors important in drug release by such routes are solubility of drug
in the surrounding tissues, molecular weight, partition coefficient and pKa
of the drug and contact surface between the drug and the surrounding tissues.
(c) Transdermal Route: Low dose
drugs like nitroglycerine can be administered by this route. The route is best suited for drugs showing extensive
first-pass metabolism upon oral administration. Important factors to be
considered for percutaneous drug absorption are partition coefficient of drug,
contact area, skin condition, skin permeability of drug, skin perfusion rate,
etc.
In short, the main determinants in deciding a route
for administration of a controlled-release system are physicochemical
properties of the drug, dose size, absorption efficiency and desired duration
of action.
B. Pharmacokinetic Characteristics of a Drug in the Design of CRDDS
A detailed knowledge of the ADME characteristics of
a drug is essential in the design of a controlled-release product. An optimum
range of a given pharmacokinetic parameter of a drug is necessary beyond which
controlled delivery is difficult or impossible.
1. Absorption Rate: For a drug to be administered as controlled-release
formulation, its absorption must be
efficient since the desired rate-limiting step is rate of drug release Kr
i.e. Kr << Ka. A drug with slow absorption is a
poor candidate for such dosage forms since continuous release will result in a
pool of unabsorbed drug e.g. iron. Aqueous soluble but poorly absorbed potent
drugs like decamethonium are also unsuitable candidates since a slight
variation in the absorption may precipitate potential toxicity.
2. Elimination Half-Life: An ideal CRDDS is the one from which rate of drug of absorption (for extended period of time) is equal
to the rate of elimination. Smaller
the t½, larger the amount
of drug to be incorporated in the controlled-release dosage form. For drugs
with t½ less than 2 hours, a very large dose may be required to
maintain the high release rate.
Drugs with half-life in the range 2 to 4 hours make good candidates for such a
system e.g. propranolol. Drugs with long half-life need not be presented in
such a formulation e.g. amlodipine. For some drugs e.g. MAO inhibitors, the
duration of action is longer than that predicted by their half-lives. A
candidate drug must have t½ that can be correlated with its pharmacological response. In terms of MRT,
a drug administered as controlled-release dosage form should have MRT
significantly longer than that from conventional dosage forms.
3. Rate of Metabolism: A drug
which is extensively metabolized is suitable for controlled-release system as long as the rate of metabolism is not
too rapid. The extent of metabolism should be identical and predictable when
the drug is administered by different routes. A drug capable of inducing or
inhibiting metabolism is a poor candidate for such a product since steady-state
blood levels would be difficult to maintain.
4. Dosage Form Index (DI): It is defined as the ratio of Css,max to Css,min. Since the goal of
controlled-release formulation is to improve therapy by reducing the dosage
form index while maintaining the plasma drug levels within the therapeutic
window, ideally its value should be as close to one as possible.
C. Pharmacodynamic Characteristics of a Drug in the Design of CRDDS
1. Drug Dose: In general, dose strength of 1.0 g is considered
maximum for a CRDDS.
2. Therapeutic Range: A candidate drug for controlled-release drug
delivery system should have a
therapeutic range wide enough such that variations in the release rate do not
result in a concentration beyond this level.
3. Therapeutic Index (TI): The release rate of a drug with narrow therapeutic index should be such that the plasma
concentration attained is within the therapeutically safe and effective range.
This is necessary because such drugs have toxic concentration nearer to their
therapeutic range. Precise control of release rate of a potent drug with narrow
margin of safety is difficult. A drug with short half-life and narrow
therapeutic index should be administered more frequently than twice a day. One
must also consider the activity of drug metabolites since controlled delivery
system controls only the release of parent drug but not its metabolism.
4. Plasma Concentration-Response (PK/PD) Relationship: Drugs such as reserpine whose
pharmacological activity is independent of its concentration are poor
candidates for controlled-release systems.
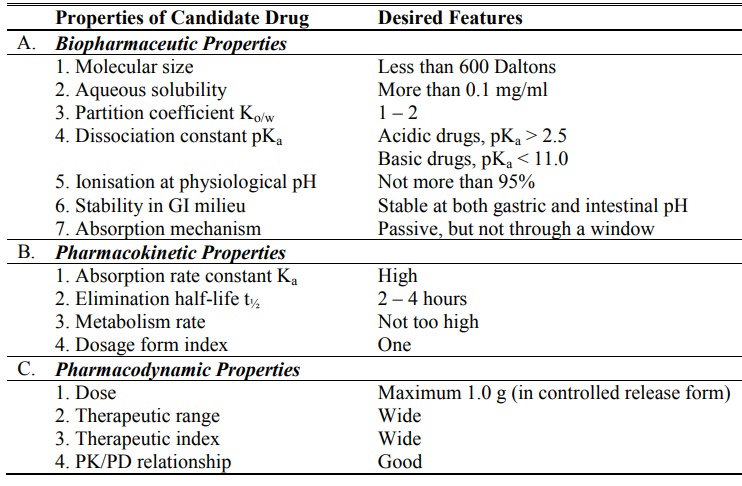
A summary of desired biopharmaceutic,
pharmacokinetic and pharmacodynamic properties of a drug is given in table
14.1.
TABLE 14.1.
Factors in the Design of CRDDS
Related Topics