Chapter Summary, Questions Answers - Amino Acids: Disposal of Nitrogen
| Home | | Biochemistry |Chapter: Biochemistry : Amino Acids: Disposal of Nitrogen
Nitrogen enters the body in a variety of compounds present in food, the most important being amino acids contained in dietary protein.
CHAPTER SUMMARY
Nitrogen enters the
body in a variety of compounds present in food, the most important being amino
acids contained in dietary protein. Nitrogen leaves the body as urea, ammonia,
and other products derived from amino acid metabolism (Figure 19.21). Free
amino acids in the body are produced by hydrolysis of dietary protein by
proteases activated from their zymogen form in the stomach and intestine,
degradation of tissue proteins, and de novo synthesis. This amino acid pool is
consumed in the synthesis of body protein, metabolized for energy, or its
members used as precursors for other nitrogen-containing compounds. Free amino
acids from digestion are taken up by intestinal cells via sodium-linked
secondary active transport. Note that body protein is simultaneously degraded
and resynthesized, a process known as protein turnover. The concentration of a
cellular protein may be determined by regulation of its synthesis or
degradation. The adenosine triphosphate (ATP)-dependent, cytosolic, selective
ubiquitin– proteasome and ATP-independent, nonselective lysosomal acid hydrolases
are the two major enzyme systems that are responsible for degrading proteins.
Nitrogen cannot be stored, and amino acids in excess of the biosynthetic needs
of the cell are quickly degraded. The first phase of catabolism involves the
transfer of the α-amino groups through transamination by
pyridoxalphosphate–dependent aminotransferases (transaminases), followed by
oxidative deamination of glutamate by glutamate dehydrogenase, forming ammonia
and the corresponding α-keto acids. A portion of the free ammonia is excreted
in the urine, some of which is used in converting glutamate to glutamine for
safe transport, but most is used in the hepatic synthesis of urea, which is
quantitatively the most important route for disposing of nitrogen from the body.
The two major causes of hyperammonemia (with its neurologic effects) are liver
disease and inherited deficiencies of urea cycle enzymes such as X-linked
ornithine transcarbamolyase.
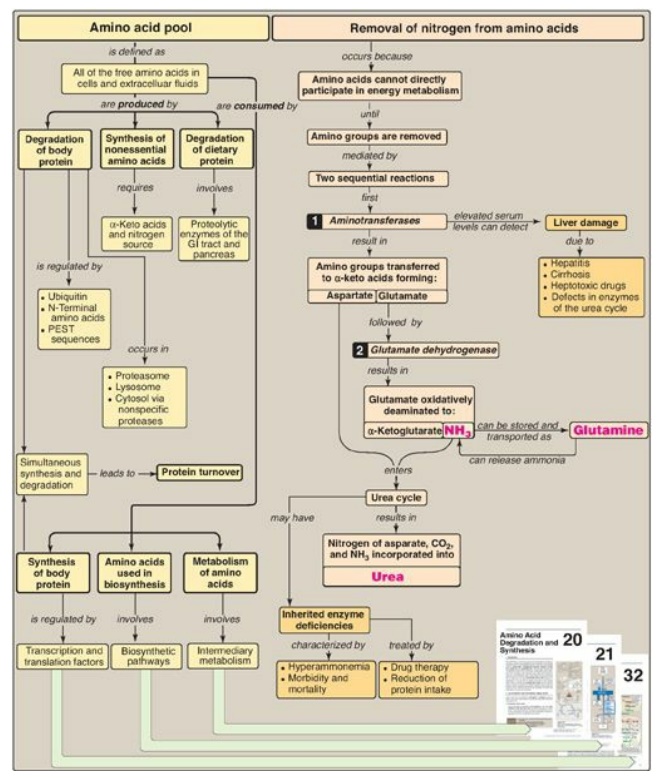
Figure 19.21 Key concept map
for nitrogen metabolism. GI = gastrointestinal; PEST = proline, glutamate,
serine, threonine; NH3 = ammonia.
Study Questions:
Choose the ONE best answer.
19.1 In the transamination reaction shown to the
right, which of the following are the products X and Y?
A. Alanine,
α-ketoglutarate
B. Asparate, α-ketoglutarate
C. Glutamate, alanine
D. Pyruvate, aspartate
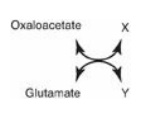
Correct answer = B. Transamination reactions always
have an amino acid and an α-keto acid as substrates. The products of the reaction
are also an amino acid (corresponding to the α-keto substrate) and an α-keto
acid (corresponding to the amino acid substrate). Three amino acid α-keto acid
pairs commonly encountered in metabolism are: alanine/pyruvate,
aspartate/oxaloacetate, and glutamate/α-ketoglutarate. In this question,
glutamate is deaminated to form α-ketoglutarate, and oxaloacetate is aminated
to form aspartate.
19.2 Which one of the following statements about
amino acids and their metabolism is correct?
A. Free amino acids are
taken into the enterocytes by a proton-linked transport system.
B. In healthy, fed
individuals, the input to the amino acid pool exceeds the output.
C. Liver uses ammonia
to buffer protons.
D. Muscle-derived glutamine is metabolized in liver
and kidney tissue to ammonia plus a gluconeogenic precursor.
E. The first step in
the catabolism of most amino acids is their oxidative deamination.
F. The toxic ammonia
generated from the amide nitrogen of amino acids is transported through blood
as arginine.
Correct answer = D. Glutamine, produced by the
catabolism of branched-chain amino acids in muscle, is deamidated to ammonia
plus glutamate. The glutamate is deaminated to ammonia plus α-ketoglutarate,
which can be used for gluconeogenesis. Free amino acids are taken into
enterocytes by a sodium-linked transport system. Healthy, fed individuals are
in nitrogen balance, in which nitrogen input equals output. Liver converts
ammonia to urea, and kidney uses ammonia to buffer protons. Amino acid
catabolism begins with transamination that generates glutamate. The glutamate
undergoes oxidative deamination. Toxic ammonia is transported as glutamine and
alanine. Arginine is synthesized and hydrolyzed in the hepatic urea cycle.
For Questions 19.3–
19.5:
A female neonate did
well until approximately age 24 hours, when she became lethargic. A sepsis
workup proved negative. At 56 hours, she started showing focal seizure
activity. The plasma ammonia level was found to be 887 µmol/l (normal 5–35
µmol/l). Quantitative plasma amino acid levels revealed a marked elevation of
citrulline but not argininosuccinate.
19.3 Which one of the following enzymic activities
is most likely to be deficient in this patient?
A. Arginase
B. Argininosuccinate
lyase
C. Argininosuccinate
synthetase
D. Carbamoyl phosphate
synthetase I
E. Ornithine
transcarbamoylase
Correct answer = C. Genetic deficiencies of each of
the five enzymes of the urea cycle, as well as deficiencies in N-acetyglutamate
synthase, have been described. The accumulation of citrulline (but not
argininosuccinate) in the plasma of this patient means that the enzyme required
for the conversion of citrulline to argininosuccinate (argininosucinate
synthetase) is defective, whereas the enzyme that cleaves argininosuccinate
(argininosuccinate lyase) is functional.
19.4 Which one of the following would also be
elevated in the blood of this patient?
A. Asparagine
B. Glutamine
C. Lysine
D. Urea
Correct answer = B. Deficiencies of the enzymes of the
urea cycle result in the failure to synthesize urea and lead to hyperammonemia
in the first few weeks after birth. Glutamine will also be elevated because it
acts as a nontoxic storage and transport form of ammonia. Therefore, elevated
glutamine always accompanies hyperammonemia. Asparagine and lysine do not serve
this sequestering role. Urea would be decreased due to impaired activity of the
urea cycle. [Note: Alanine would also be elevated in this patient.]
19.5 Why might supplementation with arginine be of
benefit to this patient?
The arginine will be
cleaved by arginase to urea and ornithine. Ornithine will be combined with
carbamoyl phosphate by ornithine transcarbamoylase to form citrulline.
Citrulline, containing one waste nitrogen, will be excreted.
Related Topics
