Digestion, Absorption, Secretion, and Utilization of Dietary Lipids
| Home | | Biochemistry |Chapter: Biochemistry : Dietary Lipid Metabolism
The average daily intake of lipids by U.S. adults is about 81 g, of which more than 90% is normally triacylglycerol ([TAG], formerly called triglyceride).
DIGESTION, ABSORPTION, SECRETION, AND UTILIZATION
OF DIETARY LIPIDS
The average daily
intake of lipids by U.S. adults is about 81 g, of which more than 90% is
normally triacylglycerol ([TAG], formerly called triglyceride). The remainder
of the dietary lipids consists primarily of cholesterol, cholesteryl esters,
phospholipids, and unesterified (“free”) fatty acids. The digestion of dietary
lipids is summarized in Figure 15.2.
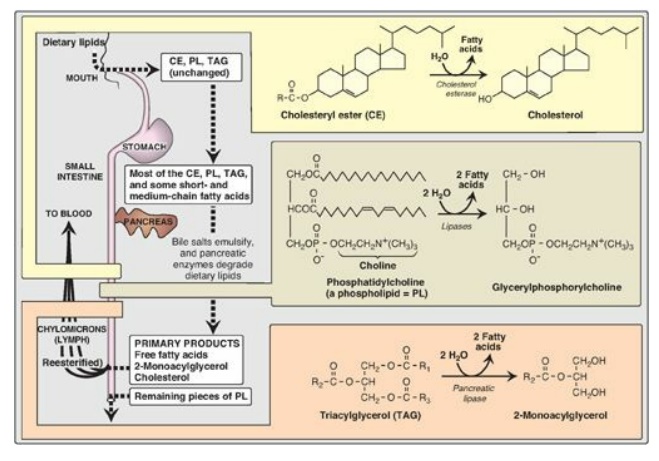
Figure 15.2 Overview of lipid digestion.
A. Processing of dietary lipid in the stomach
The digestion of lipids begins in the stomach, catalyzed by a lipase (lingual lipase) that originates from glands at the back of the tongue. TAG molecules, particularly those containing fatty acids of short- or medium-chain length (fewer than 12 carbons such as are found in milk fat), are the primary target of this enzyme. These same TAGs are also degraded by a separate gastric lipase, secreted by the gastric mucosa. Both enzymes are relatively acid stable, with pH optimums of pH 4 to pH 6. These “acid lipases” play a particularly important role in lipid digestion in neonates, for whom milk fat is the primary source of calories. They also become important digestive enzymes in individuals with pancreatic insufficiency such as those with cystic fibrosis (CF). Lingual and gastric lipases aid these patients in degrading TAG molecules (especially those with short- to medium-chain length fatty acids) despite a near or complete absence of pancreatic lipase (see below).
1. Cystic fibrosis: CF is the most common lethal
genetic disease in Caucasians of Northern European ancestry and has a
prevalence of about 1:3,300 births in the United States. CF is an autosomal
recessive disorder caused by mutations to the gene for the CF transmembrane
conductance regulator (CFTR) protein that functions as a chloride channel on
epithelium in the pancreas, lungs, testes, and sweat gands. Defective CFTR
results in decreased secretion of chloride and increased uptake of sodium and
water. In the pancreas, the depletion of water on the cell surface results in
thickened secretions that clog the pancreatic ducts, preventing pancreatic
enzymes from reaching the intestine, thereby leading to pancreatic
insufficiency. Treatment includes replacement of these enzymes and
supplementation with fat-soluble vitamins. [Note: CF also causes chronic lung
infections with progressive pulmonary disease and male infertility.]
B. Emulsification of dietary lipid in the small intestine
The critical process of
emulsification of dietary lipids occurs in the duodenum. Emulsification
increases the surface area of the hydrophobic lipid droplets so that the
digestive enzymes, which work at the interface of the droplet and the
surrounding aqueous solution, can act effectively. Emulsification is
accomplished by two complementary mechanisms, namely, use of the detergent
properties of the conjugated bile salts and mechanical mixing due to
peristalsis. Bile salts, made in the liver and stored in the gallbladder, are
amphipathic derivatives of cholesterol. Conjugated bile salts consist of a
hydroxylated sterol ring structure with a side chain to which a molecule of
glycine or taurine is covalently attached by an amide linkage (Figure 15.3).
These emulsifying agents interact with the dietary lipid particles and the
aqueous duodenal contents, thereby stabilizing the particles as they become
smaller from peristalsis and preventing them from coalescing. A more complete
discussion of bile salt metabolism.
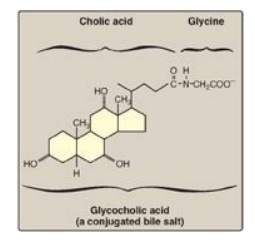
Figure 15.3 Structure of
glycocholic acid.
C. Degradation of dietary lipids by pancreatic enzymes
The dietary TAG,
cholesteryl esters, and phospholipids are enzymically degraded (“digested”) by
pancreatic enzymes, whose secretion is hormonally controlled.
1. Triacylglycerol degradation: TAG molecules are too large to be
taken up efficiently by the mucosal cells of the intestinal villi. They are,
therefore, acted upon by an esterase, pancreatic lipase, which preferentially
removes the fatty acids at carbons 1 and 3. The primary products of hydrolysis
are, thus, a mixture of 2-monoacylglycerol and free fatty acids (see Figure
15.2). [Note: This enzyme is found in high concentrations in pancreatic
secretions (2%–3% of the total protein present), and it is highly efficient
catalytically, thus insuring that only severe pancreatic deficiency, such as
that seen in CF, results in significant malabsorption of fat.] A second
protein, colipase, also secreted by the pancreas, binds the lipase at a ratio
of 1:1 and anchors it at the lipid–aqueous interface. Colipase restores
activity t o lipase in the presence of inhibitory substances like bile salts
that bind the micelles. [Note: Colipase is secreted as the zymogen,
procolipase, which is activated in the intestine by trypsin.] Orlistat, an
antiobesity drug, inhibits gastric and pancreatic lipases, thereby decreasing
fat absorption, resulting in weight loss.
2. Cholesteryl ester degradation: Most dietary cholesterol is
present in the free (nonesterified) form, with 10%–15% present in the
esterified form. Cholesteryl esters are hydrolyzed by pancreatic cholesteryl
ester hydrolase (cholesterol esterase), which produces cholesterol plus free
fatty acids (see Figure 15.2). Activity of this enzyme is greatly increased in
the presence of bile salts.
3. Phospholipid degradation: Pancreatic juice is rich in the
proenzyme of phospholipase A2 that, like procolipase, is activated
by trypsin and, like cholesteryl ester hydrolase, requires bile salts for
optimum activity. Phospholipase A2 removes one fatty acid from
carbon 2 of a phospholipid, leaving a lysophospholipid. For example,
phosphatidylcholine (the predominant phospholipid of digestion) becomes
lysophosphatidylcholine. The remaining fatty acid at carbon 1 can be removed by
lysophospholipase, leaving a glycerylphosphoryl base (for example,
glycerylphosphorylcholine, see Figure 15.2) that may be excreted in the feces,
further degraded, or absorbed.
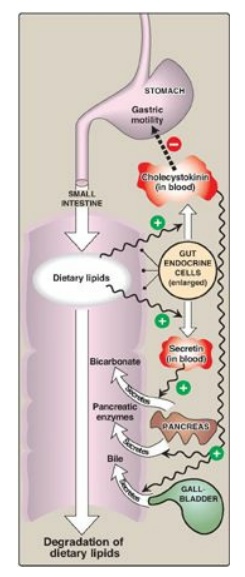
Figure 15.4 Hormonal control
of lipid digestion in the small intestine.
4. Control of lipid digestion: Pancreatic secretion of the
hydrolytic enzymes that degrade dietary lipids in the small intestine is
hormonally controlled (Figure 15.4). Cells in the mucosa of the lower duodenum
and jejunum produce a small peptide hormone, cholecystokinin (CCK), in response
to the presence of lipids and partially digested proteins entering these
regions of the upper small intestine. CCK acts on the gallbladder (causing it
to contract and release bile, a mixture of bile salts, phospholipids, and free
cholesterol) and on the exocrine cells of the pancreas (causing them to release
digestive enzymes). It also decreases gastric motility, resulting in a slower
release of gastric contents into the small intestine. Other intestinal cells
produce another small peptide hormone, secretin, in response to the low pH of
the chyme entering the intestine. Secretin causes the pancreas to release a
solution rich in bicarbonate that helps neutralize the pH of the intestinal
contents, bringing them to the appropriate pH for digestive activity by
pancreatic enzymes.
D. Absorption of lipids by intestinal mucosal cells, or enterocytes
Free fatty acids, free
cholesterol, and 2-monoacylglycerol are the primary products of lipid digestion
in the jejunum. These, plus bile salts and fat-soluble vitamins (A, D, E, and
K), form mixed micelles (that is, disc-shaped clusters of a mixture of
amphipathic lipids that coalesce with their hydrophobic groups on the inside
and their hydrophilic groups on the outside). Mixed micelles are, therefore,
soluble in the aqueous environment of the intestinal lumen (Figure 15.5). These
particles approach the primary site of lipid absorption, the brush border
membrane of the enterocytes (mucosal cell). This membrane is separated from the
liquid contents of the intestinal lumen by an unstirred water layer that mixes
poorly with the bulk fluid. The hydrophilic surface of the micelles facilitates
the transport of the hydrophobic lipids through the unstirred water layer to
the brush border membrane where they are absorbed. Bile salts are absorbed in
the terminal ileum, with less than 5% being lost in the feces. [Note: Relative
to other dietary lipids, cholesterol is only poorly absorbed by the
enterocytes. Drug therapy (for example, with ezetimibe) can further reduce
cholesterol absorption in the small intestine.] Short- and medium-chain length
fatty acids are water soluble and, thus, do not require the assistance of mixed
micelles for absorption by the intestinal mucosa.
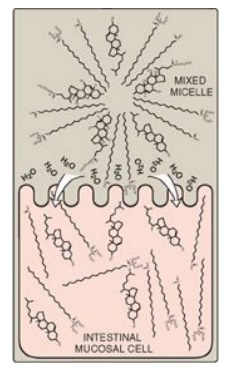
Figure 15.5 Absorption of lipids contained in a mixed micelle by an intestinal mucosal cell. [Note: The micelle itself is not taken up.]
E. Resynthesis of triacylglycerols and cholesteryl esters
The mixture of lipids
absorbed by the enterocytes migrates to the endoplasmic reticulum where
biosynthesis of complex lipids takes place. The long-chain length fatty acids
are first converted into their activated form by fatty acyl-coenzyme A (CoA)
synthetase (thiokinase) as shown in Figure 15.6. Using the fatty acyl CoA
derivatives, the 2-monoacylglycerols absorbed by the enterocytes are converted
to TAGs through sequential reacylations by two acyltransferases, acyl
CoA:monoacylglycerol acyltransferase and acyl CoA:diacylglycerol
acyltransferase. Lysophospholipids are reacylated to form phospholipids by a
family of acyltransferases, and cholesterol is esterified with a fatty acid
primarily by acyl CoA:cholesterol acyltransferase. [Note: Virtually all
long-chain fatty acids entering the enterocytes are used in this fashion to
form TAGs, phospholipids, and cholesteryl esters. Short- and medium-chain
length fatty acids are not converted to their CoA derivatives and are not
reesterified to 2-monoacylglycerol. Instead, they are released into the portal
circulation, where they are carried by serum albumin to the liver.]
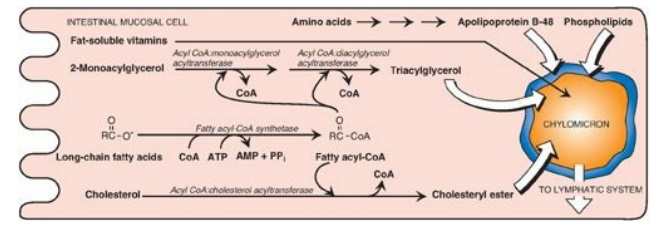
Figure 15.6 Assembly and secretion of chylomicrons by intestinal mucosal cells. [Note: Short- and medium-chain length fatty acids do not require incorporation into micelles or chylomicrons and directly enter into the blood.] CoA = coenzyme A; AMP = adenosine monophosphate; PPi = pyrophosphate.
F. Lipid malabsorption
Lipid malabsorption,
resulting in increased lipid (including the fat-soluble vitamins and essential
fatty acids,) in the feces, a condition known as steatorrhea, can be caused by
disturbances in lipid digestion and/or absorption (Figure 15.7). Such
disturbances can result from several conditions, including CF (causing poor digestion)
and short bowel syndrome (causing decreased absorption).
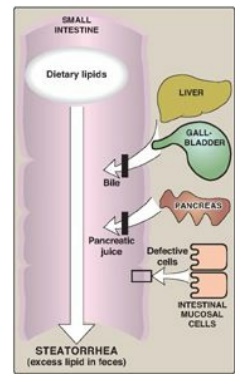
Figure 15.7 Possible causes of steatorrhea.
The ability of short- and medium-chain length fatty acids to be taken up by enterocytes without the aid of mixed micelles has made them important in dietary therapy for individuals with malabsorption disorders.
G. Secretion of lipids from enterocytes
The newly resynthesized TAGs and cholesteryl esters are very hydrophobic and aggregate in an aqueous environment. It is, therefore, necessary that they be packaged as particles of lipid droplets surrounded by a thin layer composed of phospholipids, unesterified cholesterol, and a molecule of the protein apolipoprotein B-48. This layer stabilizes the particle and increases its solubility, thereby preventing multiple particles from coalescing. [Note: Microsomal triglyceride transfer protein is essential for the assembly of these (and other) TAG-rich apolipoprotein B– containing particles in the endoplasmic reticulum.] The lipoprotein particles are released by exocytosis from enterocytes into the lacteals (lymphatic vessels originating in the villi of the small intestine). The presence of these particles in the lymph after a lipid-rich meal gives it a milky appearance. This lymph is called chyle (as opposed to chyme, the name given to the semifluid mass of partially digested food that passes from the stomach to the duodenum), and the particles are named chylomicrons. Chylomicrons follow the lymphatic system to the thoracic duct and are then conveyed to the left subclavian vein, where they enter the blood. The steps in the production of chylomicrons are summarized in Figure 15.6. [Note: Once released into blood, chylomicrons pick up apolipoproteins E and C-II.] (For a more detailed description of chylomicron structure and metabolism,)
H. Use of dietary lipids by the tissues
TAG contained in
chylomicrons is broken down primarily in the capillaries of skeletal and
cardiac muscle and adipose tissues. TAG in chylomicrons is degraded to free
fatty acids and glycerol by lipoprotein lipase (LPL). This enzyme is
synthesized primarily by adipocytes and muscle cells. It is secreted and
becomes associated with the luminal surface of endothelial cells in the
capillary beds of the peripheral tissues. [Note: Familial LPL deficiency (type
I hyperlipoproteinemia) is a rare, autosomal recessive disorder caused by a
deficiency of LPL or its coenzyme apolipoprotein C-II. The result is fasting
chylomicronemia and hypertriacylglycerolemia.]
1. Fate of free fatty acids: The free fatty acids derived from
the hydrolysis of TAG may either directly enter adjacent muscle cells or
adipocytes or be transported in the blood in association with serum albumin
until they are taken up by cells. [Note: Serum albumin is a large glycoprotein
secreted by the liver. It transports a number of primarily hydrophobic
compounds in the circulation, including free fatty acids and some drugs.] Most
cells can oxidize fatty acids to produce energy. Adipocytes can also reesterify
free fatty acids to produce TAG molecules, which are stored until the fatty
acids are needed by the body.
2. Fate of glycerol: Glycerol released from TAG is taken up from the blood and phosphorylated by hepatic glycerol kinase to produce glycerol 3-phosphate, which can enter either glycolysis or gluconeogenesis by oxidation to dihydroxyacetone phosphate.
3. Fate of the remaining chylomicron components: After most of the TAG has been removed, the chylomicron remnants (which contain cholesteryl esters, phospholipids, apolipoproteins, fat-soluble vitamins, and a small amount of TAG) bind to receptors on the liver (apolipoprotein E is the ligand;) and are endocytosed. The intracellular remnants are hydrolyzed to their component parts. Cholesterol and the nitrogenous bases of phospholipids (for example, choline) can be recycled by the body. [Note: If removal of remnants by the liver is decreased due to impaired binding to their receptor, they accumulate in the plasma. This is seen in the rare type III hyperlipoproteinemia (also called familial dysbetalipoproteinemia,).]
Related Topics
