Primary Structure of Proteins
| Home | | Biochemistry |Chapter: Biochemistry : Structure of Proteins
The sequence of amino acids in a protein is called the primary structure of the protein.
PRIMARY STRUCTURE OF PROTEINS
The sequence of amino
acids in a protein is called the primary structure of the protein.
Understanding the primary structure of proteins is important because many genetic
diseases result in proteins with abnormal amino acid sequences, which cause
improper folding and loss or impairment of normal function. If the primary
structures of the normal and the mutated proteins are known, this information
may be used to diagnose or study the disease.
A. Peptide bond
In proteins, amino
acids are joined covalently by peptide bonds, which are amide linkages between
the α-carboxyl group of one amino acid and the α-amino group of another. For
example, valine and alanine can form the dipeptide valylalanine through the
formation of a peptide bond (Figure 2.2). Peptide bonds are resistant to
conditions that denature proteins, such as heating and high concentrations of
urea. Prolonged exposure to a strong acid or base at elevated temperatures is
required to break these bonds nonenzymically.
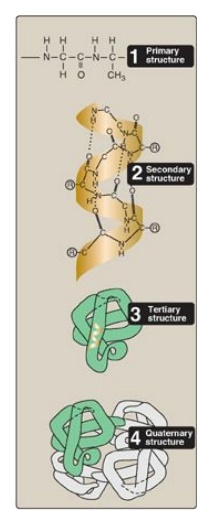
Figure 2.1
Four hierarchies of protein structure.
1. Naming the peptide: By convention, the free amino end
(N-terminal) of the peptide chain is written to the left and the free carboxyl
end (C-terminal) to the right. Therefore, all amino acid sequences are read
from the N- to the C-terminal end of the peptide. For example, in Figure 2.2A,
the order of the amino acids is “valine, alanine.” Linkage of many amino acids
through peptide bonds results in an unbranched chain called a polypeptide. Each
component amino acid in a polypeptide is called a “residue” because it is the
portion of the amino acid remaining after the atoms of water are lost in the
formation of the peptide bond. When a polypeptide is named, all amino acid
residues have their suffixes (-ine, -an, -ic, or -ate) changed to -yl, with the
exception of the C-terminal amino acid. For example, a tripeptide composed of
an N-terminal valine, a glycine, and a C-terminal leucine is called
valylglycylleucine.
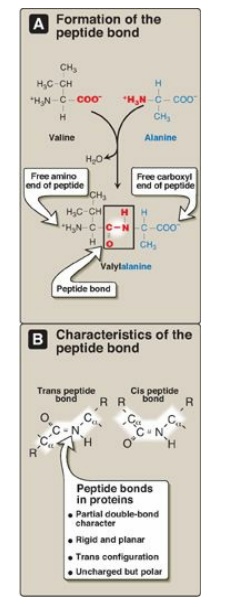
Figure 2.2 A. Formation of a peptide bond, showing the structure of the dipeptide valylalanine. B. Characteristics of the peptide bond.
2. Characteristics of the peptide bond: The peptide bond has a partial
double-bond character, that is, it is shorter than a single bond and is rigid
and planar (Figure 2.2B). This prevents free rotation around the bond between
the carbonyl carbon and the nitrogen of the peptide bond. However, the bonds
between the α-carbons and the α-amino or α-carboxyl groups can be freely
rotated (although they are limited by the size and character of the R groups).
This allows the polypeptide chain to assume a variety of possible
configurations. The peptide bond is almost always a trans bond (instead of cis,
see Figure 2.2B), in large part because of steric interference of the R groups
when in the cis position.
3. Polarity of the peptide bond: Like all amide linkages, the –ICI =O and –I NH groups of the peptide bond are uncharged and neither accept nor release protons over the pH range of 2–12. Thus, the charged groups present in polypeptides consist solely of the N-terminal (α-amino) group, the C-terminal (α-carboxyl) group, and any ionized groups present in the side chains of the constituent amino acids. The – IC=O and –I NH groups of the peptide bond are polar, however, and are involved in hydrogen bonds (for example, in α-helices and β-sheets).
B. Determination of the amino acid composition of a polypeptide
The first step in
determining the primary structure of a polypeptide is to identify and
quantitate its constituent amino acids. A purified sample of the polypeptide to
be analyzed is first hydrolyzed by strong acid at 110°C for 24 hours. This
treatment cleaves the peptide bonds and releases the individual amino acids, which
can be separated by cation-exchange chromatography. In this technique, a
mixture of amino acids is applied to a column that contains a resin to which a
negatively charged group is tightly attached. [Note: If the attached group is
positively charged, the column becomes an anion-exchange column.] The amino
acids bind to the column with different affinities, depending on their charges,
hydrophobicity, and other characteristics. Each amino acid is sequentially
released from the chromatography column by eluting with solutions of increasing
ionic strength and pH (Figure 2.3). The separated amino acids contained in the
eluate from the column are quantitated by heating them with ninhydrin (a
reagent that forms a purple compound with most amino acids, ammonia, and
amines). The amount of each amino acid is determined spectrophotometrically by
measuring the amount of light absorbed by the ninhydrin derivative. The
analysis described above is performed using an amino acid analyzer, an
automated machine whose components are depicted in Figure 2.3.
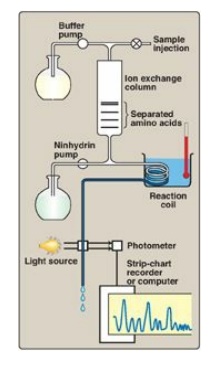
Figure 2.3
Determination of the amino acid composition of a polypeptide using an amino
acid analyzer.
C. Sequencing of the peptide from its N-terminal end
Sequencing is a
stepwise process of identifying the specific amino acid at each position in the
peptide chain, beginning at the N-terminal end. Phenylisothiocyanate, known as
Edman reagent, is used to label the amino-terminal residue under mildly
alkaline conditions (Figure 2.4). The resulting phenylthiohydantoin (PTH)
derivative introduces an instability in the N-terminal peptide bond such that
it can be hydrolyzed without cleaving the other peptide bonds. The identity of
the amino acid derivative can then be determined. Edman reagent can be applied
repeatedly to the shortened peptide obtained in each previous cycle. The
process is now automated.
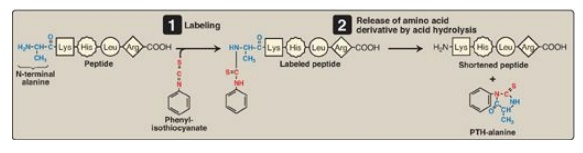
Figure 2.4 Determination of the amino (N)-terminal residue of a polypeptide by Edman degradation. PTH = phenylthiohydantoin.
D. Cleavage of the polypeptide into smaller fragments
Many polypeptides have
a primary structure composed of more than 100 amino acids. Such molecules cannot
be sequenced directly from end to end. However, these large molecules can be
cleaved at specific sites and the resulting fragments sequenced. By using more
than one cleaving agent (enzymes and/or chemicals) on separate samples of the
purified polypeptide, overlapping fragments can be generated that permit the
proper ordering of the sequenced fragments, thereby providing a complete amino
acid sequence of the large polypeptide (Figure 2.5). Enzymes that hydrolyze
peptide bonds are termed peptidases (proteases). [Note: Exopeptidases cut at
the ends of proteins and are divided into aminopeptidases and
carboxypeptidases. Carboxypeptidases are used in determining the C-terminal
amino acid. Endopeptidases cleave within a protein.]
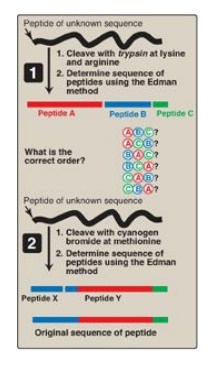
Figure 2.5
Overlapping of peptides produced by the action of trypsin and cyanogen bromide.
E. Determination of a protein’s primary structure by DNA sequencing
The sequence of
nucleotides in a protein-coding region of the DNA specifies the amino acid
sequence of a polypeptide. Therefore, if the nucleotide sequence can be
determined, it is possible, from knowledge of the genetic code, to translate
the sequence of nucleotides into the corresponding amino acid sequence of that
polypeptide. This indirect process, although routinely used to obtain the amino
acid sequences of proteins, has the limitations of not being able to predict
the positions of disulfide bonds in the folded chain and of not identifying any
amino acids that are modified after their incorporation into the polypeptide.
Therefore, direct protein sequencing is an extremely important tool for
determining the true character of the primary sequence of many polypeptides.
Related Topics
